Etnias do Alto Xingu se reúnem anualmente no Kuarup: origem dos povos nativos é mais complexa do que se sabia (foto: Mário Vilella / Funai)
Maria Guimarães | Pesquisa FAPESP – Os povos indígenas que habitam a América do Sul descendem de três ondas migratórias. A novidade é que uma delas, mais representada na população atual, veio da Mesoamérica por volta de 1.300 anos atrás, de acordo com estudo feito apenas por pesquisadores do continente. Isso revela uma maior complexidade na história dos povos nativos, com maior diversidade genética do que se antecipava. A pesquisa estampa a capa da última edição (07/05) da revista científica Nature. “Chegamos a essas conclusões por meio de um trabalho muito intenso do ponto de vista de colaborações”, conta a geneticista Tábita Hünemeier, do Instituto de Biociências da Universidade de São Paulo (IB-USP). Ela coordenou o estudo, no qual vem trabalhando há mais de uma década, e se surpreendeu com a diversidade genética mais alta do que esperava.
Foram 128 genomas sequenciados por inteiro, representando 45 povos de oito países latino-americanos – Argentina, Bolívia, Brasil, Colômbia, Equador, México, Paraguai e Peru –, e comparados a outras 71 sequências disponíveis em bancos de dados. A ideia foi estimar as afinidades genéticas entre todos os grupos indígenas americanos, levando em conta genomas antigos. A pesquisadora celebra a presença, entre os autores, da biomédica Putira Sacuena, da Universidade Federal do Pará (UFPA). “Ela foi a primeira mulher indígena a trabalhar com antropologia genética”, afirma. A colaboração indígena em estudos que dizem respeito aos povos nativos é considerada pelos pesquisadores uma novidade bem-vinda na busca por compreender essa história.
Esse trabalho acrescenta informações importantes sobre o que se sabe da colonização humana da América do Sul. A primeira onda migratória deixou registros com idades de até 12 mil anos na Lapa do Santo (leia mais em: revistapesquisa.fapesp.br/os-povos-de-lagoa-santa/) e na gruta do Sumidouro, na região mineira de Lagoa Santa, e no Chile. Por volta de 9 mil anos atrás, mais uma migração deixou marcas distintas no registro genético e arqueológico, no Peru e na Argentina. Mas o Holoceno Médio, período entre 8 mil e 4,2 mil anos atrás, trouxe mudanças ambientais que prejudicaram ecossistemas e a disponibilidade de recursos, afetando também as populações humanas.
Os povos indígenas que hoje habitam o continente, em parte por isso, descendem também de indivíduos que chegaram cerca de 1.300 anos atrás a partir de onde agora é o México. Essa terceira onda, que não estava documentada até agora, é a grande novidade. As análises do DNA indicam também que após a chegada dos europeus no século 16, os grupos indígenas se tornaram menos populosos e mais isolados uns dos outros. No tronco Tupi, o estudo detectou sinais de endocruzamento – quando a reprodução se dá entre grupos pequenos, sem possibilidades de migração – nos povos Sirionó, Suruí e Karitiana, indicando um colapso populacional provavelmente resultante de epidemias, escravização, perturbações nas possibilidades de subsistência e no conhecimento tradicional. É possível enxergar uma recuperação recente em algumas regiões da parte ocidental da América do Sul. A diversidade genética é maior na América Central e no Cone Sul.
Um enigma foi encontrar trechos genômicos muito antigos característicos da Australásia (Austrália e ilhas na região), de neandertais (da Europa) e de denisovanos (do leste asiático), preservados no DNA sul-americano. A hipótese é de que esses genes antigos tenham um papel benéfico ainda desconhecido e foram mantidos por seleção natural. O foco do artigo era a diversidade e os percursos das populações, e não os aspectos funcionais, mas a identificação de regiões associadas à resposta imune, a traços cardiometabólicos, à fertilidade e a traços antropométricos sugere que estudos futuros podem explorar mais a fundo o papel da evolução humana no continente. De acordo com Hünemeier, os marcadores genéticos usados em pesquisas anteriores tinham sido desenhados a partir de populações europeias e africanas, e não eram adequados para entender a América. “Agora temos parâmetros.”
O importante – e que contraria algumas visões sobre os grupos nativos – foi documentar a permanência prolongada de grupos humanos em muitas áreas, com uma diversidade genética pronunciada. Isso indica a necessidade de uma representação mais completa desses povos em bancos genômicos globais. “O mundo inteiro dispunha de dados genômicos para contar a história de sua população, só o Brasil não tinha”, avalia o arqueólogo André Strauss, do Museu de Arqueologia e Etnologia (MAE) da USP, que não participou do estudo. Ele remete a um artigo publicado por ele em 2018 na revista Cell, sobre a história antiga da população sul-americana (leia mais em: revistapesquisa.fapesp.br/quando-havia-indios-em-lagoa-santa/), que deixou um mistério no ar: se os povos de Lagoa Santa não eram os ancestrais diretos dos indígenas atuais, quem são esses ancestrais? “O artigo de agora confirma as duas levas migratórias anteriores e caracteriza a terceira.”
Strauss tem o objetivo de encontrar essa onda no registro arqueogenético. “Boa parte dos esqueletos que temos são mais antigos, há muito poucos dos grupos ceramistas”, explica ele. Um motivo é que as cavernas e os sambaquis são ambientes mais propícios à preservação dos esqueletos, enquanto em locais como a Amazônia eles se decompõem. Do que é possível contar a partir dos dados moleculares, há mais a caminho. “Já temos mais mil amostras sequenciadas”, afirma Hünemeier. “Entendemos que, para enxergar a diversidade da América e sua complexidade, o melhor é ter poucos indivíduos de muitas populações.”
A investigação foi apoiada pela FAPESP por meio dos projetos 15/26875-9 e 21/06860-8.
Nobody knows what around a fifth of your genes actually do. It’s hoped they could hold the secret to fixing developmental disorders, cancer, neurodegeneration, and more.
One could be forgiven for a little genetic déjà vu.
Launched in 1990, the Human Genome Project unveiled its first readout of the human DNA sequence with great fanfare in 2000. The human genome was declared essentially complete in 2003—but it took nearly 20 more years before the final, complete version was released.
This did not mark the end of humankind’s genetic puzzle, however. A new study has mapped the yawning gap between reading our genes and understanding them. Vast parts of the genome—areas the study authors have nicknamed the “Unknome”—are made of genes whose function we still don’t know.
This has important implications for medicine: Genes are the instructions for making the protein building blocks of the body. Plenty of those still shrouded in darkness could have profound medical significance and may hold the keys to disorders of development, cancer, neurodegeneration, and more.
The study makes it embarrassingly clear just how many important genes we know little to nothing about. It estimates that a fifth of human genes with a vital function are still essentially a mystery. The good news is that the research also outlines how scientists can focus on those mystery genes. “We might now be at the beginning of the end of the Unknome,” says Matthew Freeman of the Dunn School of Pathology at the University of Oxford, a coauthor of the study.
The research team used two tools to find the gaps in our knowledge. First, using the plethora of existing databases of genetic information, they compared the genetic codes of many different species to reveal genes that look roughly similar.
These riffs on a genetic theme are known as conserved genes, and even if we don’t understand what they do, we know that they must be important because nature is parsimonious and tends to use the same genetic machinery to do important jobs in different organisms. “The one thing we could be confident of is that, if important, these genes would be quite well-conserved across evolution,” says Freeman.
Once they had found similar genetic riffs in worms, humans, flies, bacteria, and other organisms, the researchers could look at what was known about the function of these clearly important genes and score them accordingly, with a high “knownness” score reflecting solid understanding.
Because so much genetic information is already available on hundreds of genomes and recorded in a standardized way, it was possible to automate this scoring process. “We then asked how many of those [conserved genes] have a score of less than one, where essentially nothing is known about them,” says Freeman. “To our surprise, two decades after the first human genome, it is still an extraordinary number.”
In all, the total number of human genes with a knownness score of 1 or less is currently 1,723 out of 19,664.
By the same token, the top 10 genes identified by the team’s rummage through genetic databases corresponded with “all the most famous genes, which is reassuring,” says Sean Munro of the Laboratory of Molecular Biology in Cambridge, a study coauthor. “We recognized every single one of them, and there are already thousands of papers about each of them.”
When it came to the substantial number that were unknown, the team conducted one more study, using the best understood (at the genetic level) organism of all: Drosophila melanogaster. These fruit flies have been the subject of research for more than a century because they are easy and inexpensive to breed, have a short life cycle, produce lots of young, and can be genetically modified in numerous ways.
The team used gene editing to dial down the use of around 300 low-scoring genes found in both humans and fruit flies. “We found that one-quarter of these unknown genes were lethal—when knocked out, they caused the flies to die, and yet nobody had ever known anything about them,” says Freeman. “Another 25 percent of them caused changes in the flies—phenotypes—that we could detect in many ways.” These genes were linked with fertility, development, locomotion, protein quality control, and resilience to stress. “That so many fundamental genes are not understood was eye-opening,” Freeman says. It’s possible that variation in these genes could have very big impacts on human health.
All of this “unknomics” information is held on a database, which the team is making available for other researchers to use to discover new biology. The next step may be to hand the data on these mystery genes and the mystery proteins they create over to AI.
DeepMind’s AlphaFold, for example, can provide important insights into what mystery proteins do, notably by revealing how they interact with other proteins, says Alex Bateman of the European Bioinformatics Institute, based near Cambridge, UK. So can cryo-EM, which is a way of producing images of large, complex molecules, he says. And a University College London team has shown a systematic way to use machine learning to figure out what proteins do in yeast.
The Unknome is unusual in that it’s a biology database that will shrink as we understand it better. The paper shows that over the past decade “we have moved from 40 percent to 20 percent of the human proteome having a certain level of unknownness,” says Bateman. However, at current progress rates, working out the function of all human protein-coding genes could take more than half a century, Freeman estimates.
The discovery that so many genes remain misunderstood reflects what is called the streetlight effect, or the drunkard’s search principle, an observational bias that occurs when people only search for something where it is easiest to look. In this case, it has caused what Freeman and Munro call a “bias in biological research toward the previously studied.”
The same goes for researchers, who tend to get funding for research in relatively well-understood areas, rather than going off into what Freeman calls the wilderness. This is why the database is so important, Munro explains—it fights back against the economics of academia, which avoids things that are very poorly understood. “There is a need for a different type of support to address these unknowns,” says Munro.
But even with the database becoming available and researchers picking through it, there will still be some knowledge blind spots. The study focused on genes that are responsible for proteins. Over the past two decades, uncharted areas of the genome have also been found to harbor the code for small RNAs—scraps of genetic material that can affect other genes, and which are critical regulators of normal development and bodily functions. There may be more “unknown unknowns” lurking in the human genome.
For now, there’s still plenty to get into, and Freeman hopes this work will encourage others to study the genetic Terra Incognita: “There’s more than enough Unknome for anyone who wants to explore genuinely new biology.”
Menos de 2% das três bilhões de letras do genoma humano são dedicados às proteínas
David Cox
17 de abril de 2023
Em abril de 2003, o sequenciamento completo do “livro da vida” codificado no genoma humano foi declarado “encerrado”, após 13 anos de trabalho. O mundo estava repleto de expectativas.
Esperava-se que o Projeto Genoma Humano, depois de consumir cerca de US$ 3 bilhões (R$ 15 bilhões), trouxesse tratamentos para doenças crônicas e esclarecesse todos os detalhes determinados geneticamente sobre as nossas vidas.
Mas, enquanto as entrevistas coletivas anunciavam o triunfo desta nova era de conhecimento biológico, o manual de instruções para a vida humana já trazia consigo uma surpresa inesperada.
A convicção que prevalecia na época era que a ampla maioria do genoma humano consistiria de instruções para a produção de proteínas — os “tijolos” que constroem todos os organismos vivos e desempenham uma imensa variedade de papéis nas nossas células e entre elas.
E, com mais de 200 tipos diferentes de células no corpo humano, parecia fazer sentido que cada uma delas precisasse dos seus próprios genes para realizar suas funções necessárias.
Acreditava-se que o surgimento de conjuntos exclusivos de proteínas fosse vital na evolução da nossa espécie e dos nossos poderes cognitivos. Afinal, somos a única espécie capaz de sequenciar o nosso próprio genoma.
Mas o que descobrimos é que menos de 2% dos três bilhões de letras do genoma humano são dedicados às proteínas. Apenas cerca de 20 mil genes codificadores de proteínas foram encontrados nas longas linhas de moléculas que compõem nossas sequências de DNA.
Os geneticistas ficaram assombrados ao descobrir que os números de genes produtores de proteínas dos seres humanos são similares a algumas das criaturas mais simples do planeta. As minhocas, por exemplo, têm cerca de 20 mil desses genes, enquanto as moscas-das-frutas têm cerca de 13 mil.
Foi assim que, do dia para a noite, o mundo científico passou a enfrentar uma verdade bastante incômoda: grande parte do nosso entendimento sobre o que nos torna seres humanos talvez estivesse errada.
“Eu me lembro da incrível surpresa”, afirma o biólogo molecular Samir Ounzain, principal executivo da companhia suíça Haya Therapeutics. A empresa procura utilizar nosso conhecimento sobre a genética humana para desenvolver novos tratamentos para doenças cardiovasculares, câncer e outras enfermidades crônicas.
“Aquele foi o momento em que as pessoas começaram a se perguntar ‘será que temos um conceito errado do que é a biologia?'”
Os 98% restantes do nosso DNA ficaram conhecidos como matéria escura, ou o genoma obscuro — uma enorme e misteriosa quantidade de letras sem propósito ou significado óbvio.
Inicialmente, alguns geneticistas sugeriram que o genoma obscuro fosse simplesmente DNA lixo, uma espécie de depósito de resíduos da evolução humana. Seriam os restos de genes partidos que deixaram de ser relevantes há muito tempo.
Mas, para outros, sempre ficou claro que o genoma obscuro seria fundamental para nosso entendimento da humanidade.
“A evolução não tem absolutamente nenhuma tolerância com o lixo”, afirma Kári Stefánsson, o principal executivo da empresa islandesa deCODE Genetics, que sequenciou mais genomas inteiros do que qualquer outra instituição em todo o mundo.
Para ele, “deve haver uma razão evolutiva para manter o tamanho do genoma”.
Duas décadas se passaram e, agora, temos os primeiros indícios da função do genoma obscuro. Aparentemente, sua função primária é regular o processo de decodificação, ou expressão, dos genes produtores de proteínas.
O genoma obscuro ajuda a controlar o comportamento dos nossos genes em resposta às pressões ambientais enfrentadas pelo nosso corpo ao longo da vida, que vão desde a alimentação até o estresse, a poluição, os exercícios e a quantidade de sono. Este campo é conhecido como epigenética.
Ounzain afirma que gosta de pensar nas proteínas como o hardware que compõe a vida. Já o genoma obscuro é o software, que processa e reage às informações externas.
Por isso, quanto mais aprendemos sobre o genoma obscuro, mais compreendemos a complexidade humana e como nos tornamos quem somos hoje.
“Se você pensar em nós enquanto espécie, somos mestres da adaptação ao ambiente em todos os níveis”, afirma Ounzain. “E essa adaptação é o processamento das informações.”
“Quando você retorna à questão sobre o que nos faz ser diferentes de uma mosca ou de uma minhoca, percebemos cada vez mais que as respostas estão no genoma obscuro”, segundo ele.
Os transposons e o nosso passado evolutivo
Quando os cientistas começaram a examinar o livro da vida, em meados dos anos 2000, uma das maiores dificuldades foi o fato de que as regiões não codificadoras de proteínas do genoma humano pareciam estar repletas de sequências de DNA repetidas, conhecidas como transposons.
Essas sequências repetitivas eram tão onipresentes que compreendiam cerca da metade do genoma em todos os mamíferos vivos.
“A própria compilação do primeiro genoma humano foi mais problemática devido à presença dessas sequências repetitivas”, afirma Jef Boeke, diretor do centro médico acadêmico chamado Projeto Matéria Escura da Universidade Langone de Nova York, nos Estados Unidos.
“Analisar simplesmente qualquer tipo de sequência é muito mais fácil quando se trata de uma sequência exclusiva.”
Inicialmente, os transposons foram ignorados pelos geneticistas. A maior parte dos estudos genéticos preferiu concentrar-se puramente no exoma — a pequena região codificadora de proteínas do genoma.
Mas, ao longo da última década, o desenvolvimento de tecnologias mais sofisticadas de sequenciamento de DNA permitiu aos geneticistas estudar o genoma obscuro com mais detalhes.
Em um desses experimentos, os pesquisadores excluíram um fragmento específico de transposon de camundongos, o que fez com que a metade dos filhotes dos animais morresse antes do nascimento. O resultado demonstra que algumas sequências de transposons podem ser fundamentais para a nossa sobrevivência.
Talvez a melhor explicação sobre o motivo da existência dos transposons no nosso genoma possa ser o fato de que eles são extremamente antigos e datam das primeiras formas de vida, segundo Boeke.
Outros cientistas sugeriram que eles provêm de vírus que invadiram o nosso DNA ao longo da história humana, antes de receberem gradualmente novas funções no corpo para que tivessem algum propósito útil.
“Na maioria das vezes, os transposons são patógenos que nos infectam e podem infectar células da linha germinal, [que são] o tipo de células que transmitimos para a geração seguinte”, afirma Dirk Hockemeyer, professor assistente de biologia celular da Universidade da Califórnia em Berkeley, nos Estados Unidos.
“Eles podem então ser herdados e gerar integração estável ao genoma”, segundo ele.
Boeke descreve o genoma obscuro como um registro fóssil vivo de alterações fundamentais no nosso DNA que ocorreram há muito tempo, na história antiga.
Uma das características mais fascinantes dos transposons é que eles podem se mover de uma parte do genoma para outra — um tipo de comportamento que gerou seu nome — criando ou revertendo mutações nos genes, às vezes com consequências extraordinárias.
O movimento de um transposon para um gene diferente pode ter sido responsável, por exemplo, pela perda da cauda na grande família dos primatas, fazendo com que a nossa espécie desenvolvesse a capacidade de andar ereta.
“Aqui você tem esse evento único que teve enorme efeito sobre a evolução, gerando toda uma linhagem de grandes primatas, incluindo a nós”, segundo Boeke.
Mas, da mesma forma que nossa crescente compreensão sobre o genoma obscuro explica cada vez mais sobre a evolução, ela pode também esclarecer o motivo do surgimento das doenças.
Ounzain ressalta que, se olharmos para os estudos de associação genômica ampla (GWAS, na sigla em inglês), que pesquisam as variações genéticas entre grandes quantidades de pessoas para identificar quais delas são relacionadas a doenças, a grande maioria das variações ligadas a doenças crônicas, como a doença de Alzheimer, diabetes e doenças cardíacas, não está nas regiões de codificação de proteínas, mas sim no genoma obscuro.
O genoma obscuro e as doenças
A ilha de Panay, nas Filipinas, é mais conhecida pelas suas cintilantes areias brancas e pelo fluxo regular de turistas. Mas este local idílico esconde um segredo trágico.
Panay abriga o maior número de casos existentes no mundo de um distúrbio dos movimentos incurável, chamado distonia-parkinsonismo ligado ao X (XDP, na sigla em inglês).
Como no mal de Parkinson, as pessoas com XDP desenvolvem uma série de sintomas que afetam sua capacidade de andar e reagir rapidamente a diversas situações.
Desde a descoberta do XDP nos anos 1970, a doença só foi diagnosticada em pessoas de ascendência filipina. Este fato permaneceu um mistério por muito tempo, até que os geneticistas descobriram que todos esses indivíduos possuem a mesma variante exclusiva de um gene chamado TAF1.
O início dos sintomas parece ser causado por um transposon no meio do gene, que é capaz de regular sua função de forma a causar prejuízo ao corpo ao longo do tempo. Acredita-se que esta variante genética tenha surgido pela primeira vez cerca de 2.000 anos atrás, antes de ser transmitida e se estabelecer na população.
“O gene TAF1 é um gene essencial, ou seja, ele é necessário para o crescimento e a multiplicação de todos os tipos de células”, afirma Boeke.
“Quando você ajusta sua expressão, você tem esse defeito muito específico, que se manifesta como uma horrível forma de parkinsonismo.”
Este é um exemplo simples de como algumas sequências de DNA do genoma obscuro podem controlar a função de diversos genes, seja ativando ou reprimindo a transformação de informações genéticas em proteínas, em resposta a indicações recebidas do ambiente.
O genoma escuro também fornece instruções para a formação de diversos tipos de moléculas, conhecidas como RNAs não codificantes. Eles podem desempenhar diversos papéis, desde ajudar a fabricar algumas proteínas, bloquear a produção de outras ou ajudar a regular a atividade genética.
“Os RNAs produzidos pelo genoma obscuro agem como os maestros da orquestra, conduzindo como o seu DNA reage ao ambiente”, explica Ounzain. E estes RNAs não codificantes, agora, são cada vez mais considerados a ligação entre o genoma obscuro e diversas doenças crônicas.
A ideia é que, se fornecermos sistematicamente os sinais errados para o genoma obscuro com o nosso estilo de vida — por exemplo, com o fumo, má alimentação e inatividade —, as moléculas de RNA produzidas por ele podem fazer com que o corpo entre em um estado de doença, alterando a atividade genética, de forma a aumentar as inflamações do corpo ou promover a morte celular.
Acredita-se que certos RNAs não codificantes podem desligar ou aumentar a atividade de um gene chamado p53, que age normalmente para evitar a formação de tumores.
Em doenças complexas, como a esquizofrenia e a depressão, todo um conjunto de RNAs não codificantes pode agir em sincronia para reduzir ou aumentar a expressão de certos genes.
Mas o nosso reconhecimento cada vez maior da importância do genoma obscuro já está trazendo novos métodos de tratamento dessas doenças.
A indústria de desenvolvimento de remédios costuma se concentrar nas proteínas, mas algumas empresas estão percebendo que pode ser mais eficaz tentar interromper os RNAs não codificantes, que controlam os genes encarregados desses processos.
No campo das vacinas contra o câncer, por exemplo, as empresas realizam sequenciamento de DNA em amostras de tumores dos pacientes para tentar identificar um alvo adequado a ser atacado pelo sistema imunológico. E a maioria dos métodos concentra-se apenas nas regiões codificantes de proteínas do genoma.
Mas a empresa alemã de biotecnologia CureVac é pioneira em um método de análise das regiões não codificantes de proteínas, na esperança de encontrar um alvo que possa interromper o câncer na fonte.
Já a empresa de Ounzain, a Haya Therapeutics, atualmente está realizando um programa de desenvolvimento de drogas dirigido a uma série de RNAs não codificantes que dirigem a formação de tecidos de cicatrização, ou fibrose, no coração — um processo que pode causar insuficiência cardíaca.
Uma das esperanças é que este método possa minimizar os efeitos colaterais decorrentes de muitos remédios de uso comum.
“O problema quando medicamos as proteínas é que existem apenas cerca de 20 mil delas no corpo e a maioria é expressa em muitas células e processos diferentes, que não têm relação com a doença”, afirma Ounzain.
“Mas a atividade do genoma obscuro é extraordinariamente específica. Existem RNAs não codificantes que regulam a fibrose apenas no coração, de forma que, ao medicá-los, temos um remédio potencialmente muito seguro”, explica ele.
O desconhecido
Paralelamente, parte desse entusiasmo precisa ser atenuada pelo fato de que, em termos de compreensão do funcionamento do genoma obscuro, apenas acabamos de arranhar a superfície.
Sabemos muito pouco sobre o que os geneticistas descrevem como regras básicas: como essas sequências não codificantes de proteínas comunicam-se para regular a atividade genética? E como exatamente essas teias complexas de interações se manifestam por longos períodos de tempo até se tornarem traços de doenças, como a neurodegeneração observada no mal de Alzheimer?
“Estamos ainda no começo”, afirma Dirk Hockemeyer. “Os próximos 15 a 20 anos ainda serão assim – [iremos] identificar comportamentos específicos em células que podem gerar doenças e, em seguida, tentar identificar as partes do genoma obscuro que podem estar envolvidas na modificação desses comportamentos. Mas, agora, temos ferramentas para nos aprofundar nisso, algo que antes não tínhamos.”
Uma dessas ferramentas é a edição genética.
Jef Boeke e sua equipe estão atualmente tentando aprender mais sobre a forma de desenvolvimento dos sintomas de XDP, reproduzindo a inserção de transposons genéticos TAF1 em camundongos.
No futuro, uma versão mais ambiciosa deste projeto poderá tentar compreender como as sequências de DNA não codificantes de proteínas regulam os genes, construindo blocos de DNA sintético a partir do zero, para transplante em células de camundongos.
“Estamos agora envolvidos em pelo menos dois projetos, usando um enorme pedaço de DNA que não faz nada e tentando instalar nele todos esses elementos”, afirma Boeke.
“Colocamos um gene ali, uma sequência não codificante em frente a ele e outra mais distante, para ver como esse gene se comporta”, explica ele. “Agora, temos todas as ferramentas para realmente construir pedaços do genoma obscuro de baixo para cima e tentar entendê-lo.”
Hockemeyer prevê que, quanto mais aprendermos, mais surpresas inesperadas o livro genético da vida continuará a nos apresentar, da mesma forma que ocorreu quando o primeiro genoma foi sequenciado, 20 anos atrás.
Para ele, “as questões são muitas. O nosso genoma ainda está evoluindo ao longo do tempo? Conseguiremos decodificá-lo totalmente?”
“Ainda estamos nesse espaço escuro em aberto que estamos explorando e existem muitas descobertas realmente fantásticas à nossa espera.”
When an illustrious person dies, the hagiography usually starts while the body is still warm. The death of biologist E.O. Wilson last December 26 was no exception to this general rule. Of course, it’s considered impolite and in bad taste to speak ill of the dead right after they leave us; it can be the worst form of talking behind someone’s back. Yet there are no firm rules about when it is okay to do so. In some cases, colleagues, journalists, and other commenters never get around to “warts and all” portraits of the departed, especially when there are inconvenient truths involved. But all too often, defenders of the deceased’s reputation take it upon themselves to police the conversation, and attack those who do want to examine the warts, especially if they do it “too soon.”
I don’t doubt that Wilson is being rightly praised for his advocacy of biodiversity conservation and his contributions to our understanding of the natural world, especially that of ants and other insects. But the inconvenient truth is that Wilson, back in 1975, gave a major boost to genetic and evolutionary explanations for human behavior when he published his massive tome, Sociobiology: The New Synthesis, to the acclaim of those convinced that biology played a bigger role in human affairs than previously appreciated, and the condemnation of those who thought it played an even lesser role.
In doing so, it has been argued, Wilson also provided considerable cover to racists who have long argued that inequities in human societies—most notably, socioeconomic differences between Blacks and whites in the United States—are due to biological differences rather than structural flaws in our society. And yet, at the time Wilson’s book was published, those who objected to his ideas—or more specifically, their application to human societies—were the ones who got accused of being politically motivated.
The first round of Wilson obituaries reflected this political bias very clearly. The “Sociobiology Wars,” as they came to be known, were treated in some obits as a kind of quaint and colorful ancient history, caricatured by one of their most memorable episodes: Anti-racist activists dumping a pitcher of water on Wilson’s head during a debate at the 1978 meeting of the American Association for the Advancement of Science.
In his obituary of Wilson for the New York Times, evolution writer Carl Zimmer gave short shrift to the critics of sociobiology, describing the Sociobiology Wars as follows:
In a letter to The New York Review of Books, some denounced sociobiology as an attempt to reinvigorate tired old theories of biological determinism — theories, they claimed, that “provided an important basis for the enactment of sterilization laws and restrictive immigration laws by the United States between 1910 and 1930 and also for the eugenics policies which led to the establishment of gas chambers in Nazi Germany.”
In her book “Defenders of the Truth” (2000), Dr. Segerstrale wrote that Dr. Wilson’s critics had shown “an astounding disregard” for what he had written, arguing that they had used “Sociobiology” as an opportunity to promote their own agendas. When Dr. Wilson attended a 1978 debate about sociobiology, protesters rushed the stage shouting, “Racist Wilson, you can’t hide, we charge you with genocide!” A woman dumped ice water on him, shouting, “Wilson, you are all wet!”
In his 1975 book Sociobiology: The New Synthesis, Ed reported a monumental survey of the wide range of animal societies, including our own. That natural selection might shape human behaviors was questioned by some. Many critics made ad hominem attacks, which were short on scientific content. Ed responded vigorously, noting that the adaptive value of animal behaviors was not in dispute, however disturbing this might be to political philosophies. During this time, someone famously threw water onto Ed at a meeting—the amount involved grows with every telling of the story. When Ed told it, it was with a twinkle and an appreciation of this unique honor.
For anyone who was not around at the time, these hagiographic accounts (please read their entire texts for support for that statement) might leave the impression that the only opponents of Wilson’s application of sociobiological thinking to human affairs were crazy left-wing activists. But the truth is that noted scientists, including Wilson’s Harvard colleagues Richard Lewontin, Ruth Hubbard, and Stephen Jay Gould, were among those who carefully examined Wilson’s ideas and found them to be in the long and sordid tradition of racial thinking about human biology. At around the same time, Harvard Medical School geneticist Jon Beckwith and others founded a Sociobiology Study Group to discuss and analyze Wilson’s book and develop a critique of his ideas, based both on solid science and the history of scientific racism.
I was around at the time, a graduate student in biology at UCLA and a member of Science for the People, the organization Beckwith and some other Wilson critics belonged to. Since most of the action was on the East Coast, especially in Boston and Cambridge, MA, I was not an active member, other than subscribing to the group’s eponymous magazine. But I did follow things closely, including the infamous water pitcher episode, and the 1976 publication of Richard Dawkins’ The Selfish Gene, which greatly expanded on the idea that humans were largely at the mercy of our genes (a conclusion that Dawkins, with limited success, has tried to refute.)
But now, barely a month after Wilson’s death and while the hagiography is still more or less in full swing, we are suddenly faced with revelations that leave little doubt Wilson was—behind the scenes, and despite his public protests—a racist, or minimally, a sympathizer of race science (which is the same thing.) The scoop goes to Science for the People magazine in its new incarnation (the publication was moribund for many years), in a February 1 article by Stacy Farina and Matthew Gibbons, a wife and husband team (Farina is an assistant professor at Howard University with a PhD in evolutionary biology, and Gibbons works in public health.)
Digging into Wilson’s letters held at the U.S. national archives, Farina and Gibbons came across a trove of correspondence between Wilson and the late scientific racist J. Philippe Rushton, who died in 2012. I will leave it to readers to look at this painfully clear article, but in my view it leaves no doubt that Wilson wholeheartedly supported, encouraged, and cheered on Rushton’s bogus and long discredited attempts to show that differences between Blacks and whites in IQ, socioeconomic status, and other measures were based on biological racial differences. There is no ambiguity here, which is making it very difficult for Wilson’s apologists to question the evidence (although they will still try.)
And it turns out that while Farina and Gibbons were working in the archives, an independent pair of historians of science, Mark Borrello of the University of Minnesota and David Sepkoski at the University of Illinois, Urbana-Champaign, were looking at the same documents and coming to the same conclusions. Their somewhat more comprehensive analysis, published on February 5 in The New York Review of Books, leaves little doubt about Wilson’s real thinking. And should it be that much of surprise? Nearly all the obituaries of Wilson emphasize his roots in Alabama and the segregated University of Alabama, and depict him as a southern gentleman scientist—without any examination of the possibility that the prejudices of growing up in the south might have left their mark on Wilson’s psyche.
This new evidence matters greatly, because over all these years the conceit of Wilson and his defenders has been that they were champions of scientific truth, and their critics were driven by politics and ideology. Indeed, the term “race realism,” used by Rushton and other scientific racists as a bludgeon against anti-racists and an attempt to depict them as cowards who cannot face what science allegedly tells them, can now clearly be seen as evidence of Wilson’s own attitudes and biases (Wilson was no shrinking violet in defending his ideas, as even the hagiographic retrospectives make clear.)
In their next to last paragraph, Borrello and Sepkoski lay out clearly what is at stake in a proper and accurate understanding of Wilson’s real legacy when it comes to his writings on sociobiology, which have been very influential in the years since:
Preserving a naively hagiographic picture of his career obscures the extent to which racist and sexist bias remains a glaring vulnerability of the science that has been built on his theories; indeed, such bias can motivate and blind scientists to deeply flawed interpretations of data. Racism in science, today, rarely announces itself with a white hood. Rather, it persists in tacit and unspoken assumptions, and hides behind claims of the inherent objectivity of scientific research.
In what follows, I would like to go back over the history of the Sociobiology Wars, and attempt to salvage—as others have tried over the years—the true history of these debates. They did not consist only of activists running around with water pitchers, a very minor part of the story, but serious and conscientious scientists trying to point out fallacies in a theory of human behavior that has left its damaging marks in today’s discourse about race and justice.
My purpose is not to do a deep dive into sociobiology and the arguments pro and con, but simply to remind readers—and alert those new to the debate—that there were serious scientific issues involved, not just left vs. right politics.
“The use and abuse of biology”
The late anthropologist Marshall Sahlins/ Elkziz/ Wikimedia Commons
In 1976, the year after Wilson’s Sociobiology was published and the same year Dawkins’ The Selfish Gene appeared, Marshall Sahlins—a major figure in anthropology who died last year—published his own contribution to this literature: The use and abuse of biology: An Anthropological Critique of Sociobiology.
It’s a slim volume, only 120 pages, but certainly not a political diatribe. Sahlins argues, in effect, that anthropology is too important and too laden with its own facts and data to be left to geneticists, evolutionary biologists, and other scientists who often know more about ants and fruit flies than about human beings. Moreover, as Sahlins points out with many examples from societies around the world, human culture is too complicated—too cultural, as it were—to be reduced to simple biology, or even complex biology.
Sahlins spends a lot of the book discussing sociobiological notions of kinship and kin selection, which have been key to the thinking of sociobiologists over the decades (Wilson developed his own spin on how natural selection was acting, which I will get to shortly.) In essence, organisms, including humans, act in such ways as to increase the likelihood that their genes will get passed on to future generations. While not all proponents of this concept endorse Dawkins’ depressing contention that genes evolved to “swarm in huge colonies, safe inside gigantic lumbering robots, sealed off from the outside world, communicating with it by tortuous indirect routes, manipulating it by remote control”—especially because the lumbering robots included us humans—the idea that human behavior can be largely explained by what is best for the replication of our genes has stuck hard in much biological thinking, even today.
(I should point out here that sociobiologists and evolutionary psychologists—the latter being sort of latter-day sociobiologists—are always quick to insist that they recognize a role for the environment, and Wilson always did so when criticized. The problem is that it’s a no-brainer that environment is involved, and this disclaimer often serves to justify returning to a focus on genes as if some sort of technicality has been dealt with.)
In his book, Sahlins provided a lot of examples of cultures, studied by anthropologists, in which kinship is not defined by those who are genetically closest, but in all kinds of other ways, including ties that have nothing to do with genealogy. In doing so, he paints a much more realistic portrait of human relationships, in which we often may be more willing to die for someone who is not genetically related to us at all than a close relative (eg, an estranged sibling or parent.)
Sahlins writes:
The reason why human social behavior is not organized by the individual maximization of genetic interest is that human beings are not socially defined by their organic qualities but in terms of symbolic attributes; and a symbol is precisely a meaningful value—such as “close kinship” or “shared blood”—which cannot be determined by the physical properties of that to which it refers.
Before leaving Sahlins, I should qualify what I say above by pointing out that he did not argue that a “political framework” should not be used in analyzing sociobiology and its weaknesses in explaining human behavior. But what he did insist on is that the politics is at its root anthropological, ie, the way we describe human societies. Thus sociobiology is itself profoundly political, he concluded:
What is inscribed in the theory of sociobiology is the entrenched ideology of Western society: the assurance of its naturalness, and the claim of its inevitability.”
There is an interesting wrinkle in Wilson’s view of how natural selection operated, however, which eventually diverged from the strict focus on kin or individual selection. Dawkins and others before him, including the British evolutionary biologist John Maynard Smith, waged a fierce war against the concept of group selection, in which natural selection is postulated to act on groups of individuals rather than individuals themselves. Wilson, however, eventually threw in his lot with advocates of “multilevel” selection (what might perhaps be called group selection lite, or kin selection heavy), particularly in collaboration with the evolutionary biologist David Sloan Wilson (no relation)—the proposition that evolution can act on both the group and individual level. The two Wilsons published, in 2007, a paper in The Quarterly Review of Biology, “Rethinking the Theoretical Foundation of Sociobiology,” which led some diehard kin selection theorists to declare that E.O. Wilson had betrayed his own cause.
Thinking and studying sociobiology
Jonathan Marks /University of North Carolina
Marshall Sahlins’ foray into the sociobiology wars was just one example of anthropologists trying to weigh in with their own insights into human behavior. One of the best critiques, in my opinion, was penned by Jonathan Marks—now an anthropologist at the University of North Carolina, Charlotte, and author of “What it means to be 98% chimpanzee” and “Why I am not a scientist”—when he was still a graduate student at the University of Arizona.
In a 1980 paper for the Arizona Anthropologist, “Sociobiology, Selfish Genes, and Human Behavior: A Bio-Cultural Critique”, Marks engaged in a witty but cogent skewering of sociobiology’s misconceptions. Among his most important criticisms, in my view, is the use by sociobiologists of what the naturalist Ernst Mayr called “beanbag genetics,” in which genes are imagined as discrete entities which code for complex behaviors such as altruism, aggression, selfishness, conformity, and other attributes. Looking at genes that way made the mathematics of calculating the effects of kin selection on evolution easier, Marks pointed out; but it has resulted in severe oversimplifications that actually obscure what is going on, especially in the evolution of human behavior (if, indeed, human behavior is something that actually genetically evolves.)
Marks wrote:
Given the knowledge that a simple behavior such as aggregation in slime molds involves the interaction of fifty genes (May 1976), one may conclude that ‘conformity’ in humans, if genetically based, would be a very formidable genetic system.
This critique, by Marks and others, was prophetic. Modern genetic research reveals that there are unlikely to be individual genes for “altruism” or other traits that geneticists have tried to mathematically model in the past, but rather a constellation of hundreds or thousands of genes involved, each one adding a tiny statistical weight to the genetic makeup of an individual—and, in the end, rendering the notion of genetic determinism for any human trait essentially meaningless. This is certainly the lesson of today’s Genome Wide Association Studies (GWAS), which often require cohorts of many thousands of subjects to detect any genetic variation at all. (For more on this, I highly recommend the writings of Eric Turkheimer, a behavior geneticist who has questioned some of the commons assumptions of his field.)
Marks again:
Sociobiology of humans, without theoretical underpinnings in ‘beanbag genetics’… is a statement of social philosophy, not science; for without genes for altruism, one cannot speak of its evolution, except in a metaphorical sense. And to accept a metaphor as literally binding is surely a breach of logic.
I recommend reading Marks’ entire paper, as well as Chapter 9 in Jon Beckwith’s memoir, Making Genes, Making Waves, “It’s the Devil in Your DNA,” a chronicle of the Sociobiology Study Group and the Sociobiology Wars which certainly corresponds to how I myself remember them. Beckwith points out that the publication of Wilson’s Sociobiology was accompanied (as his death is now) with multitudes of uncritical media stories heralding the new biological explanations for sometimes mysterious human behavior—in the New York Times, People, Cosmopolitan, Playboy, Time (a cover story), Reader’s Digest, and even House and Garden.
To try to counter these one-sided accounts, Beckwith and other critics of sociobiology argued that genetic determinism (they insisted that was what sociobiology was, even if glossed up in a more sophisticated scientific veneer) was a key principle of eugenics, Nazism, and, in our day, attempts to justify unequal treatment of different groups in employment, housing, education, and other areas of life.
And of course, sociobiology was not the end of it. Some researchers believe that evolutionary psychology is the heir to sociobiology, with its panoply of “just-so” evolutionary stories for complex human behavior; and that every few years or so there is a media frenzy over recycled theories of human racial differences (The Bell Curve, published in 1994 by Richard Herrnstein and Charles Murray, is still the subject of lively debate today; for evidence that racially motivated theories in science are again on the rise, please see Superior: The return of race science by Angela Saini.)
Jon Beckwith/ Harvard Medical School
It’s going to be interesting to see what Wilson’s defenders and apologists make of his newly revealed correspondence with Rushton. Some will no doubt insist that Wilson was simply encouraging Rushton’s right to free academic inquiry, not endorsing his racist conclusions. I think that’s going to be a hard case to make; and the inquiry into Wilson’s true views is not likely to be over. There will be other letters, hidden away in archives or in the files of his friends, which may also see the light of day.
Wilson vociferously insisted, from the 1975 publication of his famous book to pretty much the day he died, that his critics were driven by political bias, but not him. That was never a credible claim. Now, with the revelations of his personal racism, it has no credibility at all.
Suggested reading.
Beckwith, Jon. Making Genes, Making Waves: A social activist in science. (2002)
Sahlins, Marshall. The use and abuse of biology: An anthropological critique of sociobiology. (1976)
Saini, Angela. Superior: The return of race science. (2019)
Segerstrale, Ullica. Defenders of the Truth. (2000)
In addition, Jon Beckwith provided me with a detailed bibliography of papers by members of the Sociobiology Study Group and other critics:
Sociobiology: The Debate Evolves. A Special Double Issue (The Philosophical Forum: A Quarterly, vol XIII, nos 2-3, 1981-82)
Vaulting Ambition: Sociobiology and the Quest for Human Nature, by Philip Kitcher (Massachusetts Institute of Technology, 1985)
Allen, E. et al. Against Sociobiology. The New York Review of Books. pp. 182, 184-6 (Nov. 13, 1975) Reprinted in A. Caplan- . in The Sociobiology Debate. ed. by A. Caplan. Harper & Row. New York . pp. 259-264 (1978)
Alper, J.S., Beckwith, J.. Chorover, S., Hunt, J., Inouye, H., Judd, T., Lange, R.V., and Sternberg, P. The Implications of Sociobiology: Science.192:424-427 (1976).
Alper, J., Beckwith, J., and Miller, L. Sociobiology is a Political Issue. in The Sociobiology Debate. ed. by A. Caplan. Harper & Row. New York 476‑488 (l978).
Alper, J., Beckwith, J. and Egelman, E. Misusing Sociobiology. The Harvard Crimson. Nov. 19, 1979.
Beckwith, J. Triumphalism in science. (A review of The Triumph of Sociobiology, by J. Alcock., Oxford Univ. Press, 2001). American Scientist. 89:461-472 (2001).
Beckwith, J. The Political Uses of Sociobiology in the United States and Europe. The Philosophical Forum. XIII, #2, Winter, l98l, p. 3ll‑32l.
Beckwith, J. Biological Backlash: A book review of K. Bock. Human Nature and History: A Response to Sociobiology. Technology Review. Oct. l98l. p.30.
Lucía Blasco, BBC News Mundo – 20 de janeiro de 2022
América. O último continente a ser povoado pelo ser humano. Uma parte do planeta Terra desconhecida do Homo sapiens por milhares de anos.
Até que uma mudança climática — entre muitas outras coisas — permitiu ao inquieto primata pisar naquela região.
Mas como a América foi povoada?
“É uma pergunta vital que ainda não resolvemos e continuamos fazendo porque pulsa em nossa curiosidade humana”, diz à BBC News Mundo, serviço de notícias em espanhol da BBC, Lawrence C. Brody, diretor do departamento de Genômica e Sociedade do Instituto Nacional de Pesquisa do Genoma Humano (NHGRI, na sigla em inglês), nos Estados Unidos.
“Os humanos anatomicamente modernos deixaram a África há pelo menos 100 mil anos e começaram a se espalhar. E em algum momento depois de 40 mil anos, os humanos desenvolveram a tecnologia necessária para começar a explorar mais ao norte”, acrescenta Víctor Moreno, pesquisador de pós-doutorado do Centro de Geogenética da Universidade de Copenhague, na Dinamarca, à BBC News Mundo.
Há várias teorias, mas a corrente dominante atual sustenta que houve uma única migração primeiro para a Ásia, depois para a Australásia e, mais tarde, para a Europa.
A América ainda estava muito longe e, sobretudo, bastante isolada.
Os estudos sobre o DNA foram fundamentais para mapear estas migrações ancestrais.
“Nosso DNA contém um arquivo enorme da história de nossos ancestrais. Um genoma pode representar a história de muitas pessoas diferentes de uma população inteira”, afirmou à BBC News Mundo a antropóloga e geneticista americana Jennifer Raff, especialista no povoamento inicial do continente americano.
Para aprender sobre a árvore genealógica de nossos ancestrais, os cientistas sequenciam o DNA humano que ainda pode ser encontrado em fósseis e esqueletos muito antigos, razão pela qual é chamado de “DNA antigo”.
DNA antigo
As tecnologias modernas de sequenciamento tornaram possível ter acesso a fragmentos de DNA sem ter que sequenciar um genoma inteiro.
“Os antropólogos tiram conclusões gerais a partir de amostras muito, muito pequenas de DNA antigo, como dentes ou fragmentos de ossos e, mais recentemente, argila e areia. Os algoritmos nos ajudam a interpretar os dados e saber se aquele DNA está contaminado”, explicou o geneticista humano Brody.
Isso deu a eles algumas respostas sobre o povoamento da América.
“Por exemplo, descobrimos que várias populações ancestrais contribuíram para a ascendência dos povos indígenas americanos, e não apenas uma como se acreditava anteriormente”, diz Raff.
“Graças a isso, agora sabemos que o cenário do povoamento da América foi muito mais complexo do que se pensava, mas também muito mais interessante.”
Para embarcar nesta jornada fascinante, devemos começar situando-nos há aproximadamente 25 mil anos na linha do tempo.
A última era do gelo
Estamos no período do Último Máximo Glacial (LGM, na sigla em inglês), a última era do gelo conhecida na história da Terra.
“O mapa-múndi era muito diferente do atual. A maior parte da América do Norte estava coberta por uma espessa camada de gelo que tornava a região inabitável”, diz Acuña-Alonzo, antropólogo geneticista da Escola Nacional de Antropologia e História (ENAH) do México.
“As condições eram bastante difíceis. Muitos lugares eram inacessíveis e cobertos de gelo. Fazia muito frio, os humanos tinham que caçar e coletar… e não sabiam quando poderia aparecer o próximo mamute!”, acrescenta o pesquisador Víctor Moreno.
Com o avanço do período glacial, o nível dos mares do mundo foi baixando, à medida que a água era armazenada nas camadas de gelo que cobriam os continentes.
“Toda a água estava sequestrada nas geleiras”, explica Moreno.
Por causa disso, havia duas grandes geleiras que cobriam quase todo o Canadá e tornavam praticamente impossível ir para o sul.
Mas no final desse período glacial, há cerca de 12 mil anos, as camadas de gelo começaram a derreter e surgiram alguns refúgios glaciares.
“Nesses locais, as condições não eram tão terríveis e ainda eram produtivas em termos de recursos para que os humanos pudessem se alimentar”, diz Moreno.
Um desses refúgios foi a Beríngia: uma ponte de terra que emergiu do mar congelado por meio da qual as primeiras populações de humanos entraram na América, segundo acredita a maioria dos pesquisadores.
Ela se estendia do que conhecemos hoje como o Alasca até a Eurásia — e era um território seco, cheio de vegetação e fauna.
Atualmente, está submersa — por isso não é possível encontrar vestígios arqueológicos —, mas há um consenso de que os ancestrais dos indígenas americanos saíram da Sibéria em direção ao Alasca por aquele trecho de terra e ficaram isolados na Beríngia durante algum tempo.
“À medida que as péssimas condições do Último Máximo Glacial melhoravam, foram abertas certas rotas — pelo litoral e pelo interior — que teriam permitido a entrada na América a partir da região da ponte terrestre de Bering”, diz Víctor Moreno.
Mas ainda há dúvidas sobre a rota que seguiram para entrar na América, sobre quantos grupos (ou quais grupos) fizeram este caminho e quando isso aconteceu.
Quando chegaram à América?
Há duas teorias sobre quando os primeiros seres humanos chegaram à América.
As duas principais correntes são a teoria do povoamento precoce (que diz que isso ocorreu há cerca de 30 mil ou 25 mil anos) e a teoria do povoamento tardio (segundo a qual isso teria acontecido há cerca de 12 mil ou 14 mil anos).
Por muito tempo, se pensou que o povoamento foi tardio. Esta hipótese também é conhecida como “teoria clássica do povoamento da América” ou “modelo Clóvis”.
Os Clóvis, considerados em meados do século 20 a cultura indígena mais antiga da América, usavam uma técnica de entalhe de pedra bastante aprimorada para caçar a fauna gigante que existia na Idade do Gelo com ferramentas que hoje conhecemos como “pontas de clóvis”.
Fonte: Getty
Durante décadas, essas “pontas de clóvis” foram encontradas em sítios arqueológicos de cerca de 13 mil anos, espalhados por várias partes da América do Norte. Por isso, se pensava que eles foram os primeiros povoadores da América.
Mas, nos últimos anos, vários estudos genéticos refutaram essa ideia.
Embora não haja consenso, hoje há mais cientistas e arqueólogos que argumentam que a ocupação da América ocorreu muito antes do que se acreditava.
“A maioria dos cientistas e arqueólogos apoia a teoria do povoamento precoce, e não tardia, mas os pesquisadores não chegam a um acordo sobre uma data específica ou sobre que sítios arqueológicos são ‘autênticos'”, diz à BBC News Mundo Jennifer Raff.
A análise genética de populações contemporâneas e antigas foi fundamental para que a teoria do povoamento precoce ganhasse peso.
No entanto, ainda há pesquisadores — principalmente arqueólogos — que continuam a defender a teoria do povoamento tardio.
“Alguns arqueólogos são céticos a respeito dos primeiros sítios arqueológicos encontrados, sobretudo porque não aceitam os métodos de datação, as associações com a atividade humana e a estratigrafia (análise dos estratos arqueológicos) que foram reportados”, explica Acuña-Alonzo.
“A verdade é que demonstrar a antiguidade da presença humana é bastante complicado e difícil, por isso só sítios arqueológicos muito bem escavados e documentados servirão para ir mudando essas posições”, acrescenta o pesquisador.
Também segue aberto o debate sobre como os primeiros seres humanos entraram no continente depois que deixaram a ponte terrestre de Bering, ou Beríngia, mas os cientistas trabalham principalmente com duas possibilidades: uma rota marítima ou uma rota terrestre.
Teoria da via marítima
A hipótese da rota marítima está ligada à teoria do povoamento precoce e tem sido respaldada por estudos arqueológicos, linguísticos e genéticos relativamente recentes.
Segundo essa teoria dominante, os primeiros humanos teriam entrado na América margeando a costa do Pacífico, já que naquela época tão fria “o nível do mar era mais baixo, e as costas muito mais amplas. Eles não teriam conseguido atravessar grandes distâncias nem correntes marítimas que não os favorecessem”, explica o antropólogo Acuña-Alonzo.
Não sabemos a data exata, pode ser há cerca de 17 mil anos ou até mesmo 20 mil ou 30 mil anos.
Teoria da rota terrestre
Mais uma vez, não há consenso, embora menos cientistas digam que a rota foi feita por terra há cerca de 13 mil anos, coincidindo com a teoria do povoamento tardio.
“Os pesquisadores que defendem esse modelo acreditam que os primeiros humanos a chegar à América fizeram isso muito depois do Último Máximo Glacial, viajando por um corredor livre de gelo que abriu caminho nas Montanhas Rochosas canadenses enquanto as geleiras recuavam”, explica Raff.
Segundo essa teoria, os humanos teriam atravessado essa “passagem” entre as geleiras pelo interior da América do Norte e, posteriormente, se espalhado pela América do Sul.
Mas, o estudo de genomas antigos e contemporâneos, a descoberta de sítios arqueológicos pré-Clóvis e alguns estudos ambientais questionam essa teoria, por isso há mais cientistas que defendem que a travessia foi feita pelo mar.
Estas pegadas pertencem a crianças e adolescentes que viveram há pelo menos 21 mil anos. Fonte: Bournemouth University, Reino Unido
Um dos achados mais recentes foi a descoberta em setembro de 2021 de pegadas humanas em um lago do Novo México, nos Estados Unidos, com mais de 20 mil anos.
Essas pegadas sugerem que os primeiros humanos chegaram à América no auge da Última Era do Gelo e que pode ter havido grandes migrações sobre as quais ainda não sabemos muito.
A miscigenação
Mal sabemos que aparência tinham os primeiros seres humanos que chegaram à América.
Para tentar descobrir quem eram, recorremos novamente à genética.
Graças a ela sabemos que os ancestrais dos primeiros americanos se separaram de seus “primos asiáticos” quando entraram na ponte terrestre de Bering, e que se misturaram muito mais do que se supunha, sobretudo durante os últimos 10 mil anos.
Os geneticistas acreditam que houve uma miscigenação entre duas populações ancestrais humanas: os antigos paleo-siberianos e os antigos asiáticos do leste, segundo Acuña-Alonzo.
Raff diz que um desses grupos habitava o que hoje é o Sudeste Asiático. Acredita-se que esse grupo tenha contribuído majoritariamente para a ancestralidade dos primeiros seres humanos que povoaram o continente americano — especificamente, cerca de 60%, indica Víctor Moreno.
O outro ramo ancestral surgiu há cerca de 39 mil anos no que hoje é o nordeste da Sibéria.
Esses dois grupos convergiram há cerca de 25 mil a 20 mil anos atrás.
Não sabemos exatamente como isso aconteceu, mas aconteceu durante uma migração da Sibéria”
diz Raff.
“Temos muito pouca ideia. O mais provável é que tenha ocorrido em algum lugar da Sibéria, mas quão perto da ponte terrestre de Bering isso aconteceu? Quão ao norte ou quão ao sul? Isso é algo que está sendo debatido porque o apoio genético, arqueológico e antropológico que temos é escasso”, diz Víctor Moreno.
O que a genética explica é o que aconteceu a seguir: houve uma série de eventos demográficos complexos e a população, novamente, se dividiu em duas.
Um ramo, o dos antigos beríngios (por sua possível conexão com a Beríngia) não teve descendentes conhecidos. O outro, dos antigos nativos americanos, sim.
Os cientistas chegaram a essas conclusões após descobrir uma afinidade genética muito grande entre grupos ancestrais da Sibéria e populações da Eurásia Oriental.
Pesquisador analisando pegadas de mais de 20 mil anos atrás encontradas nas margens de um lago no Novo México. Fonte: Universidade de Bournemouth, Reino Unido
“Sabemos, por exemplo, que os indígenas americanos estão relacionados geneticamente às populações do nordeste da Ásia por uma série de genes que permitiram a seus ancestrais economizar energia em condições climáticas muito difíceis”, acrescenta o geneticista.
Apesar dessas descobertas, eles ainda estão tentando determinar quantos povos antigos e atuais na América têm uma conexão com a linhagem genética desses antigos nativos americanos.
“Temos que aceitar que há muitas arestas dessa pergunta para as quais ainda não temos uma resposta”, diz Raff.
Na verdade, a última descoberta no Novo México deixa outra grande incógnita no ar: a possibilidade de que as primeiras populações tenham se extinguido sem deixar descendentes, sendo “substituídas” por outros povoadores quando o corredor de gelo foi formado.
Mas ainda não se sabe se foi esse o caso ou como teria acontecido.
“Não temos escolha a não ser abraçar a incerteza. Mas, ao mesmo tempo, é emocionante saber que estamos cada vez mais perto de reconstruir essa primeira viagem à América.”
Enquanto isso, os cientistas esperam que a herança genética nos dê mais respostas sobre a última grande expansão do Homo sapiens no planeta.
Créditos
Pesquisa e reportagem: Lucía Blasco Design e infografia: Cecilia Tombesi Mapa utilizado como base: Ron Blakey, NAU – NSF Programação: Zoë Thomas, Adam Allen e Marcos Gurgel Edição: Carol Olona e Ricardo Acampora Com a colaboração de Hilda Badenes e Sally Morales Projeto liderado por Carol Olona
Research shows that a positive attitude to ageing can lead to a longer, healthier life, while negative beliefs can have hugely detrimental effects
For more than a decade, Paddy Jones has been wowing audiences across the world with her salsa dancing. She came to fame on the Spanish talent show Tú Sí Que Vales (You’re Worth It) in 2009 and has since found success in the UK, through Britain’s Got Talent; in Germany, on Das Supertalent; in Argentina, on the dancing show Bailando; and in Italy, where she performed at the Sanremo music festival in 2018 alongside the band Lo Stato Sociale.
Jones also happens to be in her mid-80s, making her the world’s oldest acrobatic salsa dancer, according to Guinness World Records. Growing up in the UK, Jones had been a keen dancer and had performed professionally before she married her husband, David, at 22 and had four children. It was only in retirement that she began dancing again – to widespread acclaim. “I don’t plead my age because I don’t feel 80 or act it,” Jones told an interviewer in 2014.
According to a wealth of research that now spans five decades, we would all do well to embrace the same attitude – since it can act as a potent elixir of life. People who see the ageing process as a potential for personal growth tend to enjoy much better health into their 70s, 80s and 90s than people who associate ageing with helplessness and decline, differences that are reflected in their cells’ biological ageing and their overall life span.
Salsa dancer Paddy Jones, centre. Photograph: Alberto Teren
Of all the claims I have investigated for my new book on the mind-body connection, the idea that our thoughts could shape our ageing and longevity was by far the most surprising. The science, however, turns out to be incredibly robust. “There’s just such a solid base of literature now,” says Prof Allyson Brothers at Colorado State University. “There are different labs in different countries using different measurements and different statistical approaches and yet the answer is always the same.”
If I could turn back time
The first hints that our thoughts and expectations could either accelerate or decelerate the ageing process came from a remarkable experiment by the psychologist Ellen Langer at Harvard University.
In 1979, she asked a group of 70- and 80-year-olds to complete various cognitive and physical tests, before taking them to a week-long retreat at a nearby monastery that had been redecorated in the style of the late 1950s. Everything at the location, from the magazines in the living room to the music playing on the radio and the films available to watch, was carefully chosen for historical accuracy.
The researchers asked the participants to live as if it were 1959. They had to write a biography of themselves for that era in the present tense and they were told to act as independently as possible. (They were discouraged from asking for help to carry their belongings to their room, for example.) The researchers also organised twice-daily discussions in which the participants had to talk about the political and sporting events of 1959 as if they were currently in progress – without talking about events since that point. The aim was to evoke their younger selves through all these associations.
To create a comparison, the researchers ran a second retreat a week later with a new set of participants. While factors such as the decor, diet and social contact remained the same, these participants were asked to reminisce about the past, without overtly acting as if they were reliving that period.
Most of the participants showed some improvements from the baseline tests to the after-retreat ones, but it was those in the first group, who had more fully immersed themselves in the world of 1959, who saw the greatest benefits. Sixty-three per cent made a significant gain on the cognitive tests, for example, compared to just 44% in the control condition. Their vision became sharper, their joints more flexible and their hands more dextrous, as some of the inflammation from their arthritis receded.
As enticing as these findings might seem, Langer’s was based on a very small sample size. Extraordinary claims need extraordinary evidence and the idea that our mindset could somehow influence our physical ageing is about as extraordinary as scientific theories come.
Becca Levy, at the Yale School of Public Health, has been leading the way to provide that proof. In one of her earliest – and most eye-catching – papers, she examined data from the Ohio Longitudinal Study of Aging and Retirement that examined more than 1,000 participants since 1975.
The participants’ average age at the start of the survey was 63 years old and soon after joining they were asked to give their views on ageing. For example, they were asked to rate their agreement with the statement: “As you get older, you are less useful”. Quite astonishingly, Levy found the average person with a more positive attitude lived on for 22.6 years after the study commenced, while the average person with poorer interpretations of ageing survived for just 15 years. That link remained even after Levy had controlled for their actual health status at the start of the survey, as well as other known risk factors, such as socioeconomic status or feelings of loneliness, which could influence longevity.
The implications of the finding are as remarkable today as they were in 2002, when the study was first published. “If a previously unidentified virus was found to diminish life expectancy by over seven years, considerable effort would probably be devoted to identifying the cause and implementing a remedy,” Levy and her colleagues wrote. “In the present case, one of the likely causes is known: societally sanctioned denigration of the aged.”
Later studies have since reinforced the link between people’s expectations and their physical ageing, while dismissing some of the more obvious – and less interesting – explanations. You might expect that people’s attitudes would reflect their decline rather than contribute to the degeneration, for example. Yet many people will endorse certain ageist beliefs, such as the idea that “old people are helpless”, long before they should have started experiencing age-related disability themselves. And Levy has found that those kinds of views, expressed in people’s mid-30s, can predict their subsequent risk of cardiovascular disease up to 38 years later.
The most recent findings suggest that age beliefs may play a key role in the development of Alzheimer’s disease. Tracking 4,765 participants over four years, the researchers found that positive expectations of ageing halved the risk of developing the disease, compared to those who saw old age as an inevitable period of decline. Astonishingly, this was even true of people who carried a harmful variant of the APOE gene, which is known to render people more susceptible to the disease. The positive mindset can counteract an inherited misfortune, protecting against the build-up of the toxic plaques and neuronal loss that characterise the disease.
How could this be?
Behaviour is undoubtedly important. If you associate age with frailty and disability, you may be less likely to exercise as you get older and that lack of activity is certainly going to increase your predisposition to many illnesses, including heart disease and Alzheimer’s.
Importantly, however, our age beliefs can also have a direct effect on our physiology. Elderly people who have been primed with negative age stereotypes tend to have higher systolic blood pressure in response to challenges, while those who have seen positive stereotypes demonstrate a more muted reaction. This makes sense: if you believe that you are frail and helpless, small difficulties will start to feel more threatening. Over the long term, this heightened stress response increases levels of the hormone cortisol and bodily inflammation, which could both raise the risk of ill health.
The consequences can even be seen within the nuclei of the individual cells, where our genetic blueprint is stored. Our genes are wrapped tightly in each cell’s chromosomes, which have tiny protective caps, called telomeres, which keep the DNA stable and stop it from becoming frayed and damaged. Telomeres tend to shorten as we age and this reduces their protective abilities and can cause the cell to malfunction. In people with negative age beliefs, that process seems to be accelerated – their cells look biologically older. In those with the positive attitudes, it is much slower – their cells look younger.
For many scientists, the link between age beliefs and long-term health and longevity is practically beyond doubt. “It’s now very well established,” says Dr David Weiss, who studies the psychology of ageing at Martin-Luther University of Halle-Wittenberg in Germany. And it has critical implications for people of all generations.
Birthday cards sent to Captain Tom Moore for his 100th birthday – many cards for older people have a less respectful tone. Photograph: Shaun Botterill/Getty Images
Our culture is saturated with messages that reinforce the damaging age beliefs. Just consider greetings cards, which commonly play on of images depicting confused and forgetful older people. “The other day, I went to buy a happy 70th birthday card for a friend and I couldn’t find a single one that wasn’t a joke,” says Martha Boudreau, the chief communications officer of AARP, a special interest group (formerly known as the American Association of Retired Persons) that focuses on the issues of over-50s.
She would like to see greater awareness – and intolerance – of age stereotypes, in much the same way that people now show greater sensitivity to sexism and racism. “Celebrities, thought leaders and influencers need to step forward,” says Boudreau.
In the meantime, we can try to rethink our perceptions of our own ageing. Various studies show that our mindsets are malleable. By learning to reject fatalistic beliefs and appreciate some of the positive changes that come with age, we may avoid the amplified stress responses that arise from exposure to negative stereotypes and we may be more motivated to exercise our bodies and minds and to embrace new challenges.
We could all, in other words, learn to live like Paddy Jones.
When I interviewed Jones, she was careful to emphasise the potential role of luck in her good health. But she agrees that many people have needlessly pessimistic views of their capabilities, over what could be their golden years, and encourages them to question the supposed limits. “If you feel there’s something you want to do, and it inspires you, try it!” she told me. “And if you find you can’t do it, then look for something else you can achieve.”
Whatever our current age, that’s surely a winning attitude that will set us up for greater health and happiness for decades to come.
This is an edited extract fromThe Expectation Effect: How your Mindset Can Transform Your Life by David Robson, published by Canongate on 6 January (£18.99).
The Three Million African Genomes (3MAG) project emerged from his work on how genetic mutations among Africans contribute to conditions like sickle-cell disease and hearing impairments.
He points out that African genes hold a wealth of genetic variation, beyond that observed by scientists in Europe and elsewhere.
“We are all African but only a small fraction of Africans moved out of Africa about 20-40,000 years ago and settled in Europe and in Asia,” he says.
Only about 2% of the human genomes that have been mapped are African
Prof Wonkam is also concerned about equity. “Too little of the knowledge and applications from genomics have benefited the global south because of inequalities in health-care systems, a small local research workforce and lack of funding,” he says.
Only about 2% of the genomes mapped globally are African, and a good proportion of these are African American. This comes from a lack of prioritising funding, policies and training infrastructure, he says, but it also means the understanding of genetic medicine as a whole is lopsided.
Studies of African genomes will also help to correct injustices, he says: “Estimates of genetic risk scores for people of African descent that predict, say, the likelihood of cardiomyopathies or schizophrenia can be unreliable or even misleading using tools that work well in Europeans.”
To address these inequities, Prof Wonkam and other scientists are talking to governments, companies and professional bodies across Africa and internationally, in order to build up capacity over the next decade to make the vision a reality.
Estimates of genetic risk scores for people of African descent can be unreliable, says Prof Wonkam
The number of three million is the minimum he expects to accurately map genetic variations across Africa. As a comparison, the UK Biobank currently aims to sequence half a million genomes in under three years, but the UK’s 68 million population is just a fraction of Africa’s 1.3 billion.
Prof Wonkam says the project will take 10 years, and will cost around $450m (£335m) per year, and says industry is already showing an interest in it.
Biotech firms say they welcome any expansion of the library of African genomes.
The Centre for Proteomic and Genomic Research (CPGR) in Cape Town works with biotech firm Artisan Biomed on a variety of diagnostic tests. The firm says it is affected by the gaps in the availability of genomic information relevant to local populations.
For example, it may find a genetic mutation in someone and not know for certain if that variation is associated with a disease, especially as a marker for that particular population.
The Centre for Proteomic and Genomic Research works with private firms to further their research
“The more information you have at that level, the better the diagnosis, treatment and eventually care can be for any individual, regardless of your ethnicity,” says Dr Lindsay Petersen, chief operations officer.
Artisan Biomed says the data it collects feeds back into CPGR’s research – allowing them to design a better diagnostic toolkit that is better suited to African populations, for instance.
“Because of the limited data sets of the African genome, it needs that hand in hand connection with research and innovation, because without that it’s just another test that has been designed for a Caucasian population that may or may not have much of an effect within the African populations,” says Dr Judith Hornby Cuff.
She says the 3MAG project would help streamline processes and improve the development of research, and perhaps one day provide cheaper, more effective and more accessible health care, particularly in the strained South African system.
Dr Aron Abera hopes his company can build labs and train staff outside South Africa
One of those hoping to take part in the 3MAG project is Dr Aron Abera, genomics scientist at Inqaba Biotech in Pretoria, which offers genetic sequencing and other services to research and industry.
The firm employs over 100 people in South Africa, Ghana, Kenya, Mali, Nigeria Senegal, Tanzania, Uganda and Zimbabwe. Currently, most of the genetics samples collected in these countries are still processed in South Africa, but Dr Abera hopes to increase the number of laboratories soon.
The gaps are not only in infrastructure, but also in staff. Over the last 20 years, Inqaba has focused on using staff and interns from the African continent – but it now has to expand its training programme as well.
Back in Cape Town, Prof Wonkam says that while the costs are huge, the project will “improve capacity in a whole range of biomedical disciplines that will equip Africa to tackle public-health challenges more equitably”.
He says: “We have to be ambitious when we are in Africa. You have so many challenges you cannot see small, you have to see big – and really big.”
Keolu Fox is an assistant professor at the University of California, San Diego, where he is affiliated with the department of anthropology, the Global Health Program, the Halıcıoğlu Data Science Institute, the Climate Action Lab, the Design Lab and the Indigenous Futures Institute. His work focuses on designing and engineering genome sequencing and editing technologies to advance precision medicine for Indigenous communities.
Wa’a Kiakahi in Keaukaha, Hawaii. Credit: Keolu Fox
I am the proud descendant of people who, at least 1,000 years ago, made one of the riskiest decisions in human history: to leave behind their homeland and set sail into the world’s largest ocean. As the first Native Hawaiian to be awarded a Ph.D. in genome sciences, I realized in graduate school that there is another possible line of evidence that can give insights into my ancestors’ voyaging history: our moʻokuʻauhau, our genome. Our ancestors’ genomes were shaped by evolutionary and cultural factors, including our migration and the ebb and flow of the Pacific Ocean. They were also shaped by the devastating history of colonialism.
Through analyzing genomes from present-day peoples, we can do incredible things like determine the approximate number of wa‘a (voyaging canoes) that arrived when my ancestors landed on the island of Hawaii or even reconstruct the genomes of some of the legendary chiefs and navigators that discovered the islands of the Pacific. And beyond these scientific and historical discoveries, genomics research can also help us understand and rectify the injustices of the past. For instance, genomics might clarify how colonialism affected things like genetic susceptibility to illness—information crucial for developing population-specific medical interventions. It can also help us reconstruct the history of land use, which might offer new evidence in court cases over disputed territories and land repatriation.
First, let’s examine what we already know from oral tradition and experimental archeology about our incredible voyaging history in the Pacific. Using complex observational science and nature as their guide, my ancestors drew on bird migration patterns, wind and weather systems, ocean currents, the turquoise glint on the bottom of a cloud reflecting a lagoon, and a complex understanding of stars, constellations and physics to find the most remote places in the world. These intrepid voyagers were the first people to launch what Kanaka Maoli (Hawaiian) master navigator Nainoa Thompson refers to as the original “moonshot.”
This unbelievably risky adventure paid off: In less than 50 generations (1,000 years), my ancestors mastered the art of sailing in both hemispheres. Traveling back and forth along an oceanic superhighway the space of Eurasia in double-hulled catamarans filled to the brim with taro, sweet potatoes, pigs and chickens, using the stars at night to navigate and other advanced techniques and technologies, iteratively perfected over time. This would be humankind’s most impressive migratory feat—no other culture in human history has covered so much distance in such a short amount of time.
The history of my voyaging ancestors and their legacy has been passed to us traditionally through our ʻōlelo (language), mo‘olelo (oral history) and hula. As a Kanaka Maoli, I have grown up knowing them: of how Maui pulled the Hawaiian Islands from the sea and how Herb Kāne, Ben Finney, Tommy Holmes, Mau Piailug and many other members of the Polynesian Voyaging Society enabled the first noninstrumental voyage from Tahiti to Hawaii in over 600 years onboard the wa‘aHōkūle‘a.
Genomes from modern Pacific Islanders have enabled us to reconstruct precise timings, paths and branching patterns, or bifurcations, of these ancient voyages, giving a refined understanding of the order in which many archipelagoes in the Pacific were settled. By working collaboratively with communities, our approach has directly challenged colonial science’s legacy of taking artifacts and genetic materials without consent. Similar tools to the new genomics have no doubt been misused in the past to justify racist and social Darwinist ends. Yet by using genetic data graciously provided by multiple communities across the Pacific, and by allowing them to shape research priorities, my colleagues and I have been able to “I ka wā mamua, ka wā ma hope,” or “walk backward into the future.”
So how can our knowledge of the genomic past allow us to walk toward this better future? Genome sequence data are not just helpful in providing refined historical information, they also help us understand and treat important contemporary matters such as population-specific disease. The time frame of these ancestors’ arrival in the Pacific, and the order in which the most remote islands in the world were settled, matters for understanding the incidence and severity among Islander populations of many complex diseases today.
Think of our genetic history as a tree, with present-day populations at the tips of branches and older ones closer to the trunk. Moving backward in time—or from the tips to the trunk—you encounter places where two branches, or populations, were descended from the same ancestor. The places where the branches split represent events in settlement histories in which two populations split, often because of a migration to a new place.
These events provide key insights into what geneticists call “founder effects” and “population bottlenecks,” which are extremely important for understanding disease susceptibility. For example, if there is a specific condition in a population at the trunk of a branching event, then populations on islands that are settled later will have a higher chance of presenting that same health condition as well. Founder populations have provided key insights into rare population-specific diseases. Some examples include Ashkenazi Jews and susceptibility to Tay-Sachs disease and Mennonite communities and susceptibility to maple syrup urine disease (MSUD).
This research also sheds important light on colonialism. As European settlers arrived in the Pacific in places such as Hawaii, Tahiti, and Aotearoa (New Zealand), they didn’t just bring the printing press, the Bible and gunpowder, they brought deadly pathogens. In the case of many Indigenous peoples, historical contact with Europeans resulted in a population collapse (a loss of approximately 80 percent of an Indigenous population’s size), mostly as a result of virgin-soilepidemics of diseases such as smallpox. From Hernán Cortés to James Cook, these bottlenecks have shaped the contemporary genetics of Indigenous peoples in ways that directly impact our susceptibility to disease.
By integrating digital sequence information (DSI) from both modern and ancient Indigenous genomes in genetic regions such as the human leukocyte antigen (HLA) system, we can observe a reduction in human genetic variation in contemporary populations, as compared with ancient ones. In this way, we can observe empirically how colonialism has shaped the genomes of modern Indigenous populations.
Today fewer than 1 percent of genome-wide association studies, which identify associations between diseases and genetic variants, and less than 5 percent of clinical trials include Indigenous peoples. We have just begun to develop mRNA vaccine-based therapies that have already shown their ability to “save the world.” Given their success and potential, why not design treatments, such as gene therapies, that are population specific and reflect the local complexity that speaks to Indigenous peoples’ unique migratory histories and experiences with colonialism?
Finally, genomics also has the potential to impact the politics of Indigenous rights and specifically how we think about the history of land stewardship and belonging. For instance, emerging genomics evidence can empirically verify who first lived on contested territories—e.g., indigenous groups could prove how many generations they arrived before colonists—which could be used in a court of law to settle land and resource repatriation claims.
Genetics gives us insights into the impact of both our peoples’ proud history of migration and the shameful legacy of colonialism. We need to encourage the use of these data to design treatments for the least, the last, the looked over and the left out, and to generate policies and legal decisions that can rectify the history of injustice. In this way, genomics can connect where we come from to where we will go. Once used to make claims about Indigenous peoples’ inferiority, today the science of the genome can be part of an Indigenous future we can all believe in.
By Rachel FrittsAug. 13, 2021 , 1:25 PM 5-7 minutes
Grizzly bears in the central coastal region of British Columbia. Michelle Valberg
The bears and Indigenous humans of coastal British Columbia have more in common than meets the eye. The two have lived side by side for millennia in this densely forested region on the west coast of Canada. But it’s the DNA that really stands out: A new analysis has found that the grizzlies here form three distinct genetic groups, and these groups align closely with the region’s three Indigenous language families.
It’s a “mind-blowing” finding that shows how cultural and biological diversity in the region are intertwined, says Jesse Popp, an Indigenous environmental scientist at the University of Guelph who was not involved with the work.
The research began purely as a genetics study. Grizzlies had recently begun to colonize islands along the coast of British Columbia, and scientists and Indigenous wildlife managers wanted to know why they were making this unprecedented move. Luckily, in 2011, the region’s five First Nations set up a collaborative “bear working group” to answer exactly that sort of question. Lauren Henson, a conservation scientist with the Raincoast Conservation Foundation, partnered with working group members from the Nuxalk, Haíɫzaqv, Kitasoo/Xai’xais, Gitga’at, and Wuikinuxv Nations to figure out which mainland grizzlies were most genetically similar to the island ones.
Henson used bear hair samples that researchers involved with the working group had collected over the course of 11 years. To get the samples, the team went to remote areas of British Columbia—some of them only accessible via helicopter—and piled up leaves and sticks, covering them with a concoction of dogfish oil or a fish-based slurry. It “smells really, really terrible to us, but is intriguing to bears,” Henson says.
The researchers then surrounded this tempting pile with a square of barbed wire, which harmlessly snagged tufts of fur—and the DNA it contains—when bears came to check out the smell. In all, the group collected samples from 147 bears over about 23,500 square kilometers—an area roughly the size of Vermont.
Henson and her colleagues then used microsatellite DNA markers—regions of the genome that change frequently compared with other sections—to determine how related the bears were to each other. The scientists found three distinct genetic groups of bears living in the study area, they report this month in Ecology and Society.
DNA analysis reveals three distinct genetic groups of grizzly bears, which align with the boundaries between Indigenous language families (gray lines). L. H. Henson et. al. Ecology and Society, 26(3): 7, 2021
But they could not find any obvious physical barriers keeping them apart. The boundaries between genetic groupings didn’t correspond to the location of waterways or especially rugged or snow-covered landscapes. It’s possible, Henson says, that the bears remain genetically distinct not because they can’t travel, but because the region is so resource-rich that they haven’t needed to do so to meet their needs.
One thing did correlate with the bears’ distribution, however: Indigenous language families. “We were looking at language maps and noticed the striking visual similarity,” Henson says. When the researchers analyzed the genetic interrelatedness of bears both within and outside the area’s three language families, they found that grizzly bears living within a language family’s boundaries were much more genetically similar to one another than to bears living outside them.
The findings don’t surprise Jenn Walkus, a Wuikinuxv scientist who co-authored the study. Growing up in a remote community called Rivers Inlet, she saw firsthand that humans and bears have a lot of the same needs in terms of space, food, and other resources. It would make sense, she says, for them to settle in the same areas—ones with a steady supply of salmon, for instance. This historic interrelatedness means Canada should manage key resources with both bears and people in mind, she says. The Wuikinuxv Nation, for example, is looking into reducing its annual salmon harvest to support the bears’ needs, she notes.
Lauren Eckert, a conservation scientist at the University of Victoria who was not involved with the study, agrees that the findings could have important implications for managing the area’s bears. It’s “fascinating” and “really shocking” work, she says. The resources that shaped grizzly bear distribution in the region clearly also shaped humans, Eckert says, “which I think reinforces the idea that local knowledge and localized management are really critical.”
At the mercy of natural selection since the dawn of life, our ancestors adapted, mated and died, passing on tiny genetic mutations that eventually made humans what we are today.
But evolution isn’t bound strictly to genes anymore, a new study suggests. Instead, human culture may be driving evolution faster than genetic mutations can work.
In this conception, evolution no longer requires genetic mutations that confer a survival advantage being passed on and becoming widespread. Instead, learned behaviors passed on through culture are the “mutations” that provide survival advantages.
This so-called cultural evolution may now shape humanity’s fate more strongly than natural selection, the researchers argue.
“When a virus attacks a species, it typically becomes immune to that virus through genetic evolution,” study co-author Zach Wood, a postdoctoral researcher in the School of Biology and Ecology at the University of Maine, told Live Science.
Such evolution works slowly, as those who are more susceptible die off and only those who survive pass on their genes.
But nowadays, humans mostly don’t need to adapt to such threats genetically. Instead, we adapt by developing vaccines and other medical interventions, which are not the results of one person’s work but rather of many people building on the accumulated “mutations” of cultural knowledge.
By developing vaccines, human culture improves its collective “immune system,” said study co-author Tim Waring, an associate professor of social-ecological systems modeling at the University of Maine.
And sometimes, cultural evolution can lead to genetic evolution. “The classic example is lactose tolerance,” Waring told Live Science. “Drinking cow’s milk began as a cultural trait that then drove the [genetic] evolution of a group of humans.”
In that case, cultural change preceded genetic change, not the other way around.
The concept of cultural evolution began with the father of evolution himself, Waring said. Charles Darwin understood that behaviors could evolve and be passed to offspring just as physical traits are, but scientists in his day believed that changes in behaviors were inherited. For example, if a mother had a trait that inclined her to teach a daughter to forage for food, she would pass on this inherited trait to her daughter. In turn, her daughter might be more likely to survive, and as a result, that trait would become more common in the population.
Waring and Wood argue in their new study, published June 2 in the journal Proceedings of the Royal Society B, that at some point in human history, culture began to wrest evolutionary control from our DNA. And now, they say, cultural change is allowing us to evolve in ways biological change alone could not.
Here’s why: Culture is group-oriented, and people in those groups talk to, learn from and imitate one another. These group behaviors allow people to pass on adaptations they learned through culture faster than genes can transmit similar survival benefits.
An individual can learn skills and information from a nearly unlimited number of people in a small amount of time and, in turn, spread that information to many others. And the more people available to learn from, the better. Large groups solve problems faster than smaller groups, and intergroup competition stimulates adaptations that might help those groups survive.
As ideas spread, cultures develop new traits.
In contrast, a person only inherits genetic information from two parents and racks up relatively few random mutations in their eggs or sperm, which takes about 20 years to be passed on to their small handful of children. That’s just a much slower pace of change.
“This theory has been a long time coming,” said Paul Smaldino, an associate professor of cognitive and information sciences at the University of California, Merced who was not affiliated with this study. “People have been working for a long time to describe how evolutionary biology interacts with culture.”
It’s possible, the researchers suggest, that the appearance of human culture represents a key evolutionary milestone.
“Their big argument is that culture is the next evolutionary transition state,” Smaldino told Live Science.
Throughout the history of life, key transition states have had huge effects on the pace and direction of evolution. The evolution of cells with DNA was a big transitional state, and then when larger cells with organelles and complex internal structures arrived, it changed the game again. Cells coalescing into plants and animals was another big sea change, as was the evolution of sex, the transition to life on land and so on.
Each of these events changed the way evolution acted, and now humans might be in the midst of yet another evolutionary transformation. We might still evolve genetically, but that may not control human survival very much anymore.
“In the very long term, we suggest that humans are evolving from individual genetic organisms to cultural groups which function as superorganisms, similar to ant colonies and beehives,” Waring said in a statement.
But genetics drives bee colonies, while the human superorganism will exist in a category all its own. What that superorganism looks like in the distant future is unclear, but it will likely take a village to figure it out.
In a new study, University of Maine researchers found that culture helps humans adapt to their environment and overcome challenges better and faster than genetics.
After conducting an extensive review of the literature and evidence of long-term human evolution, scientists Tim Waring and Zach Wood concluded that humans are experiencing a “special evolutionary transition” in which the importance of culture, such as learned knowledge, practices and skills, is surpassing the value of genes as the primary driver of human evolution.
Culture is an under-appreciated factor in human evolution, Waring says. Like genes, culture helps people adjust to their environment and meet the challenges of survival and reproduction. Culture, however, does so more effectively than genes because the transfer of knowledge is faster and more flexible than the inheritance of genes, according to Waring and Wood.
Culture is a stronger mechanism of adaptation for a couple of reasons, Waring says. It’s faster: gene transfer occurs only once a generation, while cultural practices can be rapidly learned and frequently updated. Culture is also more flexible than genes: gene transfer is rigid and limited to the genetic information of two parents, while cultural transmission is based on flexible human learning and effectively unlimited with the ability to make use of information from peers and experts far beyond parents. As a result, cultural evolution is a stronger type of adaptation than old genetics.
Waring, an associate professor of social-ecological systems modeling, and Wood, a postdoctoral research associate with the School of Biology and Ecology, have just published their findings in a literature review in the Proceedings of the Royal Society B, the flagship biological research journal of The Royal Society in London.
“This research explains why humans are such a unique species. We evolve both genetically and culturally over time, but we are slowly becoming ever more cultural and ever less genetic,” Waring says.
Culture has influenced how humans survive and evolve for millenia. According to Waring and Wood, the combination of both culture and genes has fueled several key adaptations in humans such as reduced aggression, cooperative inclinations, collaborative abilities and the capacity for social learning. Increasingly, the researchers suggest, human adaptations are steered by culture, and require genes to accommodate.
Waring and Wood say culture is also special in one important way: it is strongly group-oriented. Factors like conformity, social identity and shared norms and institutions — factors that have no genetic equivalent — make cultural evolution very group-oriented, according to researchers. Therefore, competition between culturally organized groups propels adaptations such as new cooperative norms and social systems that help groups survive better together.
According to researchers, “culturally organized groups appear to solve adaptive problems more readily than individuals, through the compounding value of social learning and cultural transmission in groups.” Cultural adaptations may also occur faster in larger groups than in small ones.
With groups primarily driving culture and culture now fueling human evolution more than genetics, Waring and Wood found that evolution itself has become more group-oriented.
“In the very long term, we suggest that humans are evolving from individual genetic organisms to cultural groups which function as superorganisms, similar to ant colonies and beehives,” Waring says. “The ‘society as organism’ metaphor is not so metaphorical after all. This insight can help society better understand how individuals can fit into a well-organized and mutually beneficial system. Take the coronavirus pandemic, for example. An effective national epidemic response program is truly a national immune system, and we can therefore learn directly from how immune systems work to improve our COVID response.”
###
Waring is a member of the Cultural Evolution Society, an international research network that studies the evolution of culture in all species. He applies cultural evolution to the study of sustainability in social-ecological systems and cooperation in organizational evolution.
Wood works in the UMaine Evolutionary Applications Laboratory managed by Michael Kinnison, a professor of evolutionary applications. His research focuses on eco-evolutionary dynamics, particularly rapid evolution during trophic cascades.
In a race to cure his daughter, a Google programmer enters the world of hyper-personalized drugs.
Erika Check Hayden
February 26, 2020
To create atipeksen, Yu borrowed from recent biotech successes like gene therapy. Some new drugs, including cancer therapies, treat disease by directly manipulating genetic information inside a patient’s cells. Now doctors like Yu find they can alter those treatments as if they were digital programs. Change the code, reprogram the drug, and there’s a chance of treating many genetic diseases, even those as unusual as Ipek’s.
The new strategy could in theory help millions of people living with rare diseases, the vast majority of which are caused by genetic typos and have no treatment. US regulators say last year they fielded more than 80 requests to allow genetic treatments for individuals or very small groups, and that they may take steps to make tailor-made medicines easier to try. New technologies, including custom gene-editing treatments using CRISPR, are coming next.
Where it had taken decades for Ionis to perfect its drug, Yu now set a record: it took only eight months for Yu to make milasen, try it on animals, and convince the US Food and Drug Administration to let him inject it into Mila’s spine.
“I never thought we would be in a position to even contemplate trying to help these patients,” says Stanley Crooke, a biotechnology entrepreneur and founder of Ionis Pharmaceuticals, based in Carlsbad, California. “It’s an astonishing moment.”
Antisense drug
Right now, though, insurance companies won’t pay for individualized gene drugs, and no company is making them (though some plan to). Only a few patients have ever gotten them, usually after heroic feats of arm-twisting and fundraising. And it’s no mistake that programmers like Mehmet Kuzu, who works on data privacy, are among the first to pursue individualized drugs. “As computer scientists, they get it. This is all code,” says Ethan Perlstein, chief scientific officer at the Christopher and Dana Reeve Foundation.
A nonprofit, the A-T Children’s Project, funded most of the cost of designing and making Ipek’s drug. For Brad Margus, who created the foundation in 1993 after his two sons were diagnosed with A-T, the change between then and now couldn’t be more dramatic. “We’ve raised so much money, we’ve funded so much research, but it’s so frustrating that the biology just kept getting more and more complex,” he says. “Now, we’re suddenly presented with this opportunity to just fix the problem at its source.”
Ipek was only a few months old when her father began looking for a cure. A geneticist friend sent him a paper describing a possible treatment for her exact form of A-T, and Kuzu flew from Sunnyvale, California, to Los Angeles to meet the scientists behind the research. But they said no one had tried the drug in people: “We need many more years to make this happen,” they told him.
Timothy Yu, of Boston Children’s HospitalCourtesy Photo (Yu)
Kuzu didn’t have years. After he returned from Los Angeles, Margus handed him a thumb drive with a video of a talk by Yu, a doctor at Boston Children’s Hospital, who described how he planned to treat a young girl with Batten disease (a different neurodegenerative condition) in what press reports would later dub “a stunning illustration of personalized genomic medicine.” Kuzu realized Yu was using the very same gene technology the Los Angeles scientists had dismissed as a pipe dream.
That technology is called “antisense.” Inside a cell, DNA encodes information to make proteins. Between the DNA and the protein, though, come messenger molecules called RNA that ferry the gene information out of the nucleus. Think of antisense as mirror-image molecules that stick to specific RNA messages, letter for letter, blocking them from being made into proteins. It’s possible to silence a gene this way, and sometimes to overcome errors, too.
Though the first antisense drugs appeared 20 years ago, the concept achieved its first blockbuster success only in 2016. That’s when a drug called nusinersen, made by Ionis, was approved to treat children with spinal muscular atrophy, a genetic disease that would otherwise kill them by their second birthday.
Yu, a specialist in gene sequencing, had not worked with antisense before, but once he’d identified the genetic error causing Batten disease in his young patient, Mila Makovec, it became apparent to him he didn’t have to stop there. If he knew the gene error, why not create a gene drug? “All of a sudden a lightbulb went off,” Yu says. “Couldn’t one try to reverse this? It was such an appealing idea, and such a simple idea, that we basically just found ourselves unable to let that go.”
Yu admits it was bold to suggest his idea to Mila’s mother, Julia Vitarello. But he was not starting from scratch. In a demonstration of how modular biotech drugs may become, he based milasen on the same chemistry backbone as the Ionis drug, except he made Mila’s particular mutation the genetic target. Where it had taken decades for Ionis to perfect a drug, Yu now set a record: it took only eight months for him to make milasen, try it on animals, and convince the US Food and Drug Administration to let him inject it into Mila’s spine.
“What’s different now is that someone like Tim Yu can develop a drug with no prior familiarity with this technology,” says Art Krieg, chief scientific officer at Checkmate Pharmaceuticals, based in Cambridge, Massachusetts.
Source code
As word got out about milasen, Yu heard from more than a hundred families asking for his help. That’s put the Boston doctor in a tough position. Yu has plans to try antisense to treat a dozen kids with different diseases, but he knows it’s not the right approach for everyone, and he’s still learning which diseases might be most amenable. And nothing is ever simple—or cheap. Each new version of a drug can behave differently and requires costly safety tests in animals.
Kuzu had the advantage that the Los Angeles researchers had already shown antisense might work. What’s more, Margus agreed that the A-T Children’s Project would help fund the research. But it wouldn’t be fair to make the treatment just for Ipek if the foundation was paying for it. So Margus and Yu decided to test antisense drugs in the cells of three young A-T patients, including Ipek. Whichever kid’s cells responded best would get picked.
Ipek may not survive past her 20s without treatment.Matthew Monteith
While he waited for the test results, Kuzu raised about $200,000 from friends and coworkers at Google. One day, an email landed in his in-box from another Google employee who was fundraising to help a sick child. As he read it, Kuzu felt a jolt of recognition: his coworker, Jennifer Seth, was also working with Yu.
Seth’s daughter Lydia was born in December 2018. The baby, with beautiful chubby cheeks, carries a mutation that causes seizures and may lead to severe disabilities. Seth’s husband Rohan, a well-connected Silicon Valley entrepreneur, refers to the problem as a “tiny random mutation” in her “source code.” The Seths have raised more than $2 million, much of it from co-workers.
Custom drug
By then, Yu was ready to give Kuzu the good news: Ipek’s cells had responded the best. So last September the family packed up and moved from California to Cambridge, Massachusetts, so Ipek could start getting atipeksen. The toddler got her first dose this January, under general anesthesia, through a lumbar puncture into her spine.
After a year, the Kuzus hope to learn whether or not the drug is helping. Doctors will track her brain volume and measure biomarkers in Ipek’s cerebrospinal fluid as a readout of how her disease is progressing. And a team at Johns Hopkins will help compare her movements with those of other kids, both with and without A-T, to observe whether the expected disease symptoms are delayed.
One serious challenge facing gene drugs for individuals is that short of a healing miracle, it may ultimately be impossible to be sure they really work. That’s because the speed with which diseases like A-T progress can vary widely from person to person. Proving a drug is effective, or revealing that it’s a dud, almost always requires collecting data from many patients, not just one. “It’s important for parents who are ready to pay anything, try anything, to appreciate that experimental treatments often don’t work,” says Holly Fernandez Lynch, a lawyer and ethicist at the University of Pennsylvania. “There are risks. Trying one could foreclose other options and even hasten death.”
Kuzu says his family weighed the risks and benefits. “Since this is the first time for this kind of drug, we were a little scared,” he says. But, he concluded, “there’s nothing else to do. This is the only thing that might give hope to us and the other families.”
Another obstacle to ultra-personal drugs is that insurance won’t pay for them. And so far, pharmaceutical companies aren’t interested either. They prioritize drugs that can be sold thousands of times, but as far as anyone knows, Ipek is the only person alive with her exact mutation. That leaves families facing extraordinary financial demands that only the wealthy, lucky, or well connected can meet. Developing Ipek’s treatment has already cost $1.9 million, Margus estimates.
Some scientists think agencies such as the US National Institutes of Health should help fund the research, and will press their case at a meeting in Bethesda, Maryland, in April. Help could also come from the Food and Drug Administration, which is developing guidelines that may speed the work of doctors like Yu. The agency will receive updates on Mila and other patients if any of them experience severe side effects.
The FDA is also considering giving doctors more leeway to modify genetic drugs to try in new patients without securing new permissions each time. Peter Marks, director of the FDA’s Center for Biologics Evaluation and Research, likens traditional drug manufacturing to factories that mass-produce identical T-shirts. But, he points out, it’s now possible to order an individual basic T-shirt embroidered with a company logo. So drug manufacturing could become more customized too, Marks believes.
Custom drugs carrying exactly the message a sick kid’s body needs? If we get there, credit will go to companies like Ionis that developed the new types of gene medicine. But it should also go to the Kuzus—and to Brad Margus, Rohan Seth, Julia Vitarello, and all the other parents who are trying save their kids. In doing so, they are turning hyper-personalized medicine into reality.
Erika Check Hayden is director of the science communication program at the University of California, Santa Cruz.
As a species, humans have populated almost every corner of the earth. We have developed technologies and cultures which shape the world we live in.
The idea of ‘natural selection’ or ‘survival of the fittest’ seems to make sense in Stone Age times when we were fighting over scraps of meat, but does it still apply now?
We asked 12 experts whether humans are still evolving. The expert consensus is unanimously ‘yes’, however scientists say we might have the wrong idea of what evolution actually is.
Evolution is not the same as natural selection
Evolution is often used interchangeable with the phrases ‘survival of the fittest’ or ‘natural selection’. Actually, these are not quite the same thing.
‘Evolution’ simply means the gradual change of a population over time.
‘Natural selection’ is a mechanism by which evolution can occur. Our Stone Age ancestors who were faster runners avoided being trampled by mammoths and were more likely to have children. That is ‘natural selection’.
Overtime, the human population became faster at running. That’s evolution.
Evolution can happen without natural selection
That makes sense for Stone Age humans, but what about nowadays? We don’t need to outrun mammoths, we have medicines for when we’re sick and we can go to the shops to get food.
Natural selection needs a ‘selection pressure’ (e.g. dangerous trampling mammoths), so if we don’t have these anymore, does this mean we stop evolving?
Even with no selection pressures, experts say evolution still occurs by other mechanisms.
Professor Stanley Ambrose, an anthropologist from the University of Illinois, explains that “any change in the proportions of genes or gene variants over time is also considered evolution. The variants may be functionally equivalent, so evolution does not automatically equate with ‘improvement'”.
Whilst some genes can be affected by natural selection (e.g. genes that help us run faster), other changes in our DNA might have no obvious effect on us. ‘Neutral’ variations can also spread through a population by a different mechanism called ‘genetic drift’.
Genetic drift works by chance: some individuals might be unlucky and die for reasons which have nothing to do with their genes. Their unique gene variations will not be passed on to the next generation, and so the population will change.
Genetic drift doesn’t need any selection pressures, and it is still happening today.
Natural selection is still happening in humans
As much as we have made things easier for ourselves, there are still selection pressures around us, which mean that natural selection is still happening.
Like all mammals, humans lose the ability to digest milk when they stop breastfeeding. This is because we stop making an enzyme called lactase. In some countries, the population has acquired ‘lactase persistence’, meaning that people make lactase throughout their lives.
In European countries we can thank one specific gene variation for our lactase persistence, which is called ‘-13910*T’. By studying this specific gene variation in modern and ancient DNA samples, researchers suggest that it became common after humans started domesticated and milking animals.
This is an example of natural selection where we have actually made the selection pressure ourselves – we started drinking milk, so we evolved to digest it!
Another example of humans undergoing natural selection to adapt to a lifestyle is the Bajau people, who traditionally live in houseboats in the waters of South East Asia and spend much of their lives diving to hunt fish or collect shellfish.
There are always selective pressures around us, even ones that we create ourselves.
As Dr Benjamin Hunt from the University of Birmingham puts it, “Our technological and cultural changes alter the strength and composition of the selection pressures within our environment, but selection pressures still exist.”
Evolution can’t be stopped
So, evolution can happen by different mechanisms like natural selection and genetic drift. As our environment is always changing, natural selection is always happening. And even if our environment was ‘just right’ for us, we would evolve anyway!
Dr Alywyn Scally, an expert in evolution and genetics from the University of Cambridge, explains: “As long as human reproduction involves randomness and genetic mutation (and the laws of the Universe pretty much guarantee that this will always be the case at some level), there will continue to be differences from one generation to the next, meaning that the process of evolution can never be truly halted.”
Takeaway: Evolution means change in a population. That includes both easy-to-spot changes to adapt to an environment as well as more subtle, genetic changes.
Humans are still evolving, and that is unlikely to change in the future.
An infrared microscope image shows mosquito larvae with red-glowing eyes, part of an experiment using CRISPR gene-editing technology. MediaNews Group/Orange County Register via Getty Images
Usually good for a conspiracy theory or two, U.S. President Donald Trump has suggested that the virus causing COVID-19 was either intentionally engineered or resulted from a lab accident at the Wuhan Institute of Virology in China. Its release could conceivably have involved an accident, but the pathogen isn’t the mishmash of known viruses that one would expect from something designed in a lab, as a research report in Nature Medicine conclusively lays out. “If someone were seeking to engineer a new coronavirus as a pathogen, they would have constructed it from the backbone of a virus known to cause illness,” the researchers said.
But if genetic engineering wasn’t behind this pandemic, it could very well unleash the next one. With COVID-19 bringing Western economies to their knees, all the world’s dictators now know that pathogens can be as destructive as nuclear missiles. What’s even more worrying is that it no longer takes a sprawling government lab to engineer a virus. Thanks to a technological revolution in genetic engineering, all the tools needed to create a virus have become so cheap, simple, and readily available that any rogue scientist or college-age biohacker can use them, creating an even greater threat. Experiments that could once only have been carried out behind the protected walls of government and corporate labs can now practically be done on the kitchen table with equipment found on Amazon. Genetic engineering—with all its potential for good and bad—has become democratized.
To design a virus, a bio researcher’s first step is to obtain the genetic information of an existing pathogen—such as one of the coronaviruses that cause the common cold—which could then be altered to create something more dangerous. In the 1970s, the first genetic sequencing of a bacterium, Escherichia coli, took weeks of effort and cost millions of dollars just to determine its 5,836 base pairs, the building blocks of genetic information. Today, sequencing the 3,000,000,000 base pairs that make up the human genome, which dictates the construction and maintenance of a human being, can be done in a few hours for about $1000 in the United States. Xun Xu, the CEO of Chinese genomics research company BGI Group, told me by email that he expects to offer full human-genome sequencing in supermarkets and online for about $290 by the end of this year.
The next step in engineering a virus is to modify the genome of the existing pathogen to change its effects. One technology in particular makes it almost as easy to engineer life forms as it is to edit Microsoft Word documents. CRISPR gene editing, developed only a few years ago, deploys the same natural mechanism that bacteria use to trim pieces of genetic information from one genome and insert it into another. This mechanism, which bacteria developed over millennia to defend themselves from viruses, has been turned into a cheap, simple, and fast way to edit the DNA of any organism in the lab.
If experimenting with DNA once required years of experience, sophisticated labs, and millions of dollars, CRISPR has changed all that. To set up a CRISPR editing capability, the experimenter need only order a fragment of RNA and purchase off-the-shelf chemicals and enzymes, costing only a few dollars, on the Internet. Because it’s so cheap and easy to use, thousands of scientists all over the world are experimenting with CRISPR-based gene editing projects. Very little of this research is limited by regulations, largely because regulators don’t yet understand what has suddenly become possible.
China, with its emphasis on technological progress ahead of safety and ethics, has made the most astonishing breakthroughs. In 2014, Chinese scientists announced they had successfully produced monkeys that had been genetically modified at the embryonic stage. In April 2015, another group of researchers in China detailed the first ever-effort to edit the genes of a human embryo. While the attempt failed, it shocked the world: This wasn’t supposed to happen so soon.
In April 2016, yet another group of Chinese researchers reported having succeeded in modifying the genome of a human embryo in an effort to make it resistant to HIV infection, though the embryo was not brought to term. But then, in November 2018, Chinese researcher He Jiankui announced that he had created the first “CRISPR babies”—healthy infants whose genomes were edited before they were born. The People’s Daily gushed over the “historical breakthrough,” but after a global uproar, the Chinese authorities—who, He claims, had supported his efforts—arrested and later sentenced him to three years in prison for unethical conduct. But the Rubicon of biomedical science had been crossed.
China’s legion of rogue scientists is certainly a worry. But gene-editing technology has become so accessible that we could conceivably see teenagers experimenting with viruses. In the United States, anyone who wants to start modifying the genome in their garage can order a do-it-yourself CRISPR kit online for $169, for example. This comes with “everything you need to make precision genome edits in bacteria at home.” For $349, the same company is also offering a human engineering kit, which comes with embryonic kidney cells from a tissue culture originally taken from an aborted female human fetus. Shipment is advertised to take no longer than three days—no special couriers or ice packs needed.
Mail-order DNA fragments enabled a team at the University of Alberta, in 2017, to resurrect an extinct relative of the smallpox virus, horsepox, from scratch by stitching together the fragments. Horsepox is not known to harm humans, but experts warned that the same method could be used by scientists without much specialized knowledge to recreate smallpox—a horrific virus finally eradicated in 1980—within six months at a cost of about $100,000. Had the Canadian scientists used CRISPR, their cost would have been reduced to a fraction.
In my book, The Driver in the Driverless Car, published before the Canadian horsepox resurrection and the Chinese gene-edited babies, I warned about the dangers of gene editing, predicting we would have to make difficult choices about whether to restrict synthetic biology technologies. When used for good purposes, these technologies can help solve the problems of humanity—by quickly finding cures for diseases, for example. When used for evil, they can wreak global havoc of exactly the kind we are now fighting. That is why many people, myself included, have advocated for a moratorium on human gene editing.
But not just a moratorium: There should have been international treaties to prevent the use of CRISPR for gene editing on humans or animals. The U.S. Food and Drug Administration should have kept companies from selling DIY gene-editing kits. Governments should have placed restrictions on labs such as the University of Alberta’s. But none of this happened, nor were there any other checks and balances. It is now too late to stop the global spread of these technologies—the genie is out of the bottle.
Now, the only solution is to accelerate the good side of these technologies while building our defenses. As we are seeing with the development of vaccines for COVID-19, this is possible. In the past, vaccines took decades to create. Now, we are on track to have them within months, thanks to advances in genetic engineering. The Moderna Therapeutics and Pfizer/BioNTech vaccines, which are now in third-stage clinical trials, took only weeks to develop. It is conceivable that this could be reduced to hours once the technologies are perfected.
We can also accelerate the process of testing vaccines and treatments, which has become the slowest part of the development cycle. To test greater numbers of potential cancer drugs more quickly, for example, labs all over the world are creating three-dimensional cell cultures called “patient-derived organoids” from tumor biopsies. The leading company in this field, SEngine Precision Medicine, is able to test more than 100 drugs on these organoids, removing the need to use human subjects as the guinea pigs. Researchers at Harvard University’s Wyss Institute announced in January 2020 that they had developed the first human “organ-on-a-chip” model of the lung that accurately replicates a human organ’s physiology and pathophysiology. Engineers at the Massachusetts Institute of Technology have been developing a microfluidic platform that connects engineered tissues from up to 10 organs, allowing the replication of human-organ interactions for weeks at a time in order to measure the effects of drugs on different parts of the body. Many more such systems are being developed that could accelerate testing and treatment. All these technologies will greatly strengthen our biodefense.
There really is no turning back to correct the mistakes of the past. The genie cannot be put back in the bottle. We must treat the coronavirus pandemic as a full dress rehearsal of what is to come—unfortunately, that includes not only viruses that erupt from nature, but also those that will be deliberately engineered by humans. We must learn very quickly to build the same types of types of defenses that our computers have against their invaders. The good that might ultimately come from this is the cure for all disease. The bad is just about too terrible to think about.
This drawing of the Liverpool slave ship Brooks was commissioned by abolitionists to depict the inhumanity of the slave trade by showing how Africans were crammed below decks.
(CNN) Much of what we know about the horrors of slavery in the Americas comes from historical records. But new research shows that evidence of the slave trade’s atrocities can also be found in the DNA of African Americans.
A study conducted by the consumer genetics company 23andMe, published Thursday in theAmerican Journal of Human Genetics, offers some new insight into the consequences of the trans-Atlantic slave trade, from the scale at which enslaved Black women were raped by their White masters to the less-documented slave trade that occurred within the Americas.
It’s one of the largest studies of its kind, thanks in part to the massive database of 23andMe customers that researchers were able to recruit consenting participants from.
The authors compiled genetic data from more than 50,000 people from the Americas, Western Europe and Atlantic Africa, and compared it against the historical records of where enslaved people were taken from and where they were enslaved. Together, the data and records tell a story about the complicated roots of the African diaspora in the Americas.
For the most part, the DNA was consistent with what the documents show. But, the study authors said, there were some notable differences.
Here’s some of what they found, and what it reveals about the history of slavery.
It shows the legacy of rape against enslaved women
The enslaved workers who were taken from Africa and brought to the Americas were disproportionately male. Yet, genetic data shows that enslaved women contributed to gene pools at a higher rate.
In the US and parts of the Caribbean colonized by the British, African women contributed to the gene pool about 1.5 to 2 times more than African men. In Latin America, that rate was even higher. Enslaved women contributed to the gene pool in Central America, the Latin Caribbean and parts of South America about 13 to 17 times more.
To the extent that people of African descent in the Americas had European ancestry, they were more likely to have White fathers in their lineage than White mothers in all regions except the Latin Caribbean and Central America.
What that suggests: The biases in the gene pool toward enslaved African women and European men signals generations of rape and sexual exploitation against enslaved women at the hands of White owners, authors Steven Micheletti and Joanna Mountain wrote in an email to CNN.
That enslaved Black women were often raped by their masters “is not a surprise” to any Black person living in the US, says Ravi Perry, a political science professor at Howard University. Numerous historical accounts confirm this reality, as the study’s authors note.
But the regional differences between the US and Latin America are what’s striking.
The US and other former British colonies generally forced enslaved people to have children in order to maintain workforces — which could explain why the children of an enslaved woman were more likely to have an enslaved father. Segregation in the US could also be a factor, the authors theorized.
By contrast, the researchers point to the presence of racial whitening policies in several Latin American countries, which brought in European immigrants with the aim of diluting the African race. Such policies, as well as higher mortality rates of enslaved men, could explain the disproportionate contributions to the gene pool by enslaved women, the authors wrote.
It sheds light on the intra-American slave trade
Far more people in the US and Latin America have Nigerian ancestry than expected, given what historical records show about the enslaved people that embarked from ports along present-day Nigeria into the Americas, according to the study.
What that suggests: This is most likely a reflection of the intercolonial slave trade that occurred largely from the British Caribbean to other parts of the Americas between 1619 and 1807, Micheletti and Mountain wrote.
Once enslaved Africans arrived in the Americas, many were put on new ships and transported to other regions.”
Documented intra-American voyages indicate that the vast majority of enslaved people were transported from the British Caribbean to other parts of the Americas, presumably to maintain the slave economy as transatlantic slave trading was increasingly prohibited,” the authors wrote in the study.
When enslaved people from Nigeria who came into the British Caribbean were traded into other areas, their ancestry spread to regions that didn’t directly trade with that part of Africa.
It shows the dire conditions enslaved people faced
Conversely, ancestry from the region of Senegal and the Gambia is underrepresented given the proportion of enslaved people who embarked from there, Micheletti and Mountain said.
The reasons for that are grim.
What that suggests: One possible explanation the authors gave for the low prevalence of Senegambian ancestry is that over time, more and more children from the region were forced onto ships to make the journey to the Americas.
The unsanitary conditions in the holds of the ship led to malnourishment and illness, the authors wrote, meaning that less of them survived.
Another possibility is the dangerous conditions that enslaved people from the region faced once they arrived. A significant proportion of Senegambians were taken to rice plantations in the US, which were often rampant with malaria, Micheletti and Mountain said.
The study has limitations
The 23andMe study is significant in how it juxtaposes genetic data with historical records, as well as in the size of its dataset, experts who weren’t involved in the study told CNN.
“I’m not aware of anyone that has done such a comprehensive job of putting these things together, by a long shot,” said Simon Gravel, a human genetics professor at McGill University. “It’s really big progress.”
Still, he said, the research has its limitations.
In order to conduct their analysis, the scientists had to make “a lot of simplifications,” Gravel said. The researchers broke down African ancestry into four corresponding regions on the continent’s Atlantic Coast: Nigerian, Senegambian, Coastal West African and Congolese.”
That doesn’t tell you the whole story,” Gravel added, though he said more data is needed in the broader field of genomics for the researchers to drill down deeper.
Jada Benn Torres, a genetic anthropologist at Vanderbilt University, also said she would have liked to see a higher proportion of people from Africa included in the study. Out of the more than 50,000 participants, about 2,000 were from Africa. “
From the perspective of human evolutionary genetics, Africa is the most genetic diverse continent,” she wrote in an email to CNN. “In order to adequate capture existing variation, the sample sizes must be large.”
But both Gravel and Benn Torres called the study an exciting start that offers more information about the descendents of enslaved Africans.
And that, the researchers, said was what they set out to do.”
We hope this paper helps people in the Americas of African descent further understand where their ancestors came from and what they overcame,” Micheletti wrote.
“… To me, this is the point, to make a personal connection with the millions of people whose ancestors were forced from Africa into the Americas and to not forget what their ancestors had to endure.”
HUMANS ARE lucky to live a hundred years. Oak trees may live a thousand; mayflies, in their adult form, a single day. But they are all alive in the same way. They are made up of cells which embody flows of energy and stores of information. Their metabolisms make use of that energy, be it from sunlight or food, to build new molecules and break down old ones, using mechanisms described in the genes they inherited and may, or may not, pass on.
It is this endlessly repeated, never quite perfect reproduction which explains why oak trees, humans, and every other plant, fungus or single-celled organism you have ever seen or felt the presence of are all alive in the same way. It is the most fundamental of all family resemblances. Go far enough up any creature’s family tree and you will find an ancestor that sits in your family tree, too. Travel further and you will find what scientists call the last universal common ancestor, LUCA. It was not the first living thing. But it was the one which set the template for the life that exists today.
And then there are viruses. In viruses the link between metabolism and genes that binds together all life to which you are related, from bacteria to blue whales, is broken. Viral genes have no cells, no bodies, no metabolism of their own. The tiny particles, “virions”, in which those genes come packaged—the dot-studded disks of coronaviruses, the sinister, sinuous windings of Ebola, the bacteriophages with their science-fiction landing-legs that prey on microbes—are entirely inanimate. An individual animal, or plant, embodies and maintains the restless metabolism that made it. A virion is just an arrangement of matter.
The virus is not the virion. The virus is a process, not a thing. It is truly alive only in the cells of others, a virtual organism running on borrowed hardware to produce more copies of its genome. Some bide their time, letting the cell they share the life of live on. Others immediately set about producing enough virions to split their hosts from stem to stern.
The virus has no plan or desire. The simplest purposes of the simplest life—to maintain the difference between what is inside the cell and what is outside, to move towards one chemical or away from another—are entirely beyond it. It copies itself in whatever way it does simply because it has copied itself that way before, in other cells, in other hosts.
That is why, asked whether viruses are alive, Eckard Wimmer, a chemist and biologist who works at the State University of New York, Stony Brook, offers a yes-and-no. Viruses, he says, “alternate between nonliving and living phases”. He should know. In 2002 he became the first person in the world to take an array of nonliving chemicals and build a virion from scratch—a virion which was then able to get itself reproduced by infecting cells.
The fact that viruses have only a tenuous claim to being alive, though, hardly reduces their impact on things which are indubitably so. No other biological entities are as ubiquitous, and few as consequential. The number of copies of their genes to be found on Earth is beyond astronomical. There are hundreds of billions of stars in the Milky Way galaxy and a couple of trillion galaxies in the observable universe. The virions in the surface waters of any smallish sea handily outnumber all the stars in all the skies that science could ever speak of.
Back on Earth, viruses kill more living things than any other type of predator. They shape the balance of species in ecosystems ranging from those of the open ocean to that of the human bowel. They spur evolution, driving natural selection and allowing the swapping of genes.
They may have been responsible for some of the most important events in the history of life, from the appearance of complex multicellular organisms to the emergence of DNA as a preferred genetic material. The legacy they have left in the human genome helps produce placentas and may shape the development of the brain. For scientists seeking to understand life’s origin, they offer a route into the past separate from the one mapped by humans, oak trees and their kin. For scientists wanting to reprogram cells and mend metabolisms they offer inspiration—and powerful tools.
II A lifestyle for genes
THE IDEA of a last universal common ancestor provides a plausible and helpful, if incomplete, answer to where humans, oak trees and their ilk come from. There is no such answer for viruses. Being a virus is not something which provides you with a place in a vast, coherent family tree. It is more like a lifestyle—a way of being which different genes have discovered independently at different times. Some viral lineages seem to have begun quite recently. Others have roots that comfortably predate LUCA itself.
Disparate origins are matched by disparate architectures for information storage and retrieval. In eukaryotes—creatures, like humans, mushrooms and kelp, with complex cells—as in their simpler relatives, the bacteria and archaea, the genes that describe proteins are written in double-stranded DNA. When a particular protein is to be made, the DNA sequence of the relevant gene acts as a template for the creation of a complementary molecule made from another nucleic acid, RNA. This messenger RNA (mRNA) is what the cellular machinery tasked with translating genetic information into proteins uses in order to do so.
Because they, too, need to have proteins made to their specifications, viruses also need to produce mRNAs. But they are not restricted to using double-stranded DNA as a template. Viruses store their genes in a number of different ways, all of which require a different mechanism to produce mRNAs. In the early 1970s David Baltimore, one of the great figures of molecular biology, used these different approaches to divide the realm of viruses into seven separate classes (see diagram).
In four of these seven classes the viruses store their genes not in DNA but in RNA. Those of Baltimore group three use double strands of RNA. In Baltimore groups four and five the RNA is single-stranded; in group four the genome can be used directly as an mRNA; in group five it is the template from which mRNA must be made. In group six—the retroviruses, which include HIV—the viral RNA is copied into DNA, which then provides a template for mRNAs.
Because uninfected cells only ever make RNA on the basis of a DNA template, RNA-based viruses need distinctive molecular mechanisms those cells lack. Those mechanisms provide medicine with targets for antiviral attacks. Many drugs against HIV take aim at the system that makes DNA copies of RNA templates. Remdesivir (Veklury), a drug which stymies the mechanism that the simpler RNA viruses use to recreate their RNA genomes, was originally developed to treat hepatitis C (group four) and subsequently tried against the Ebola virus (group five). It is now being used against SARS–CoV-2 (group four), the covid-19 virus.
Studies of the gene for that RNA-copying mechanism, RdRp, reveal just how confusing virus genealogy can be. Some viruses in groups three, four and five seem, on the basis of their RdRp-gene sequence, more closely related to members of one of the other groups than they are to all the other members of their own group. This may mean that quite closely related viruses can differ in the way they store their genomes; it may mean that the viruses concerned have swapped their RdRp genes. When two viruses infect the same cell at the same time such swaps are more or less compulsory. They are, among other things, one of the mechanisms by which viruses native to one species become able to infect another.
How do genes take on the viral lifestyle in the first place? There are two plausible mechanisms. Previously free-living creatures could give up metabolising and become parasitic, using other creatures’ cells as their reproductive stage. Alternatively genes allowed a certain amount of independence within one creature could have evolved the means to get into other creatures.
Living creatures contain various apparently independent bits of nucleic acid with an interest in reproducing themselves. The smallest, found exclusively in plants, are tiny rings of RNA called viroids, just a few hundred genetic letters long. Viroids replicate by hijacking a host enzyme that normally makes mRNAs. Once attached to a viroid ring, the enzyme whizzes round and round it, unable to stop, turning out a new copy of the viroid with each lap.
Viroids describe no proteins and do no good. Plasmids—somewhat larger loops of nucleic acid found in bacteria—do contain genes, and the proteins they describe can be useful to their hosts. Plasmids are sometimes, therefore, regarded as detached parts of a bacteria’s genome. But that detachment provides a degree of autonomy. Plasmids can migrate between bacterial cells, not always of the same species. When they do so they can take genetic traits such as antibiotic resistance from their old host to their new one.
Recently, some plasmids have been implicated in what looks like a progression to true virus-hood. A genetic analysis by Mart Krupovic of the Pasteur Institute suggests that the Circular Rep-Encoding Single-Strand-DNA (CRESS–DNA) viruses, which infect bacteria, evolved from plasmids. He thinks that a DNA copy of the genes that another virus uses to create its virions, copied into a plasmid by chance, provided it with a way out of the cell. The analysis strongly suggests that CRESS–DNA viruses, previously seen as a pretty closely related group, have arisen from plasmids this way on three different occasions.
Such jailbreaks have probably been going on since very early on in the history of life. As soon as they began to metabolise, the first proto-organisms would have constituted a niche in which other parasitic creatures could have lived. And biology abhors a vacuum. No niche goes unfilled if it is fillable.
It is widely believed that much of the evolutionary period between the origin of life and the advent of LUCA was spent in an “RNA world”—one in which that versatile substance both stored information, as DNA now does, and catalysed chemical reactions, as proteins now do. Set alongside the fact that some viruses use RNA as a storage medium today, this strongly suggests that the first to adopt the viral lifestyle did so too. Patrick Forterre, an evolutionary biologist at the Pasteur Institute with a particular interest in viruses (and the man who first popularised the term LUCA) thinks that the “RNA world” was not just rife with viruses. He also thinks they may have brought about its end.
The difference between DNA and RNA is not large: just a small change to one of the “letters” used to store genetic information and a minor modification to the backbone to which these letters are stuck. And DNA is a more stable molecule in which to store lots of information. But that is in part because DNA is inert. An RNA-world organism which rewrote its genes into DNA would cripple its metabolism, because to do so would be to lose the catalytic properties its RNA provided.
An RNA-world virus, having no metabolism of its own to undermine, would have had no such constraints if shifting to DNA offered an advantage. Dr Forterre suggests that this advantage may have lain in DNA’s imperviousness to attack. Host organisms today have all sorts of mechanisms for cutting up viral nucleic acids they don’t like the look of—mechanisms which biotechnologists have been borrowing since the 1970s, most recently in the form of tools based on a bacterial defence called CRISPR. There is no reason to imagine that the RNA-world predecessors of today’s cells did not have similar shears at their disposal. And a virus that made the leap to DNA would have been impervious to their blades.
Genes and the mechanisms they describe pass between viruses and hosts, as between viruses and viruses, all the time. Once some viruses had evolved ways of writing and copying DNA, their hosts would have been able to purloin them in order to make back-up copies of their RNA molecules. And so what began as a way of protecting viral genomes would have become the way life stores all its genes—except for those of some recalcitrant, contrary viruses.
III The scythes of the seas
IT IS A general principle in biology that, although in terms of individual numbers herbivores outnumber carnivores, in terms of the number of species carnivores outnumber herbivores. Viruses, however, outnumber everything else in every way possible.
This makes sense. Though viruses can induce host behaviours that help them spread—such as coughing—an inert virion boasts no behaviour of its own that helps it stalk its prey. It infects only that which it comes into contact with. This is a clear invitation to flood the zone. In 1999 Roger Hendrix, a virologist, suggested that a good rule of thumb might be ten virions for every living individual creature (the overwhelming majority of which are single-celled bacteria and archaea). Estimates of the number of such creatures on the planet come out in the region of 1029-1030. If the whole Earth were broken up into pebbles, and each of those pebbles smashed into tens of thousands of specks of grit, you would still have fewer pieces of grit than the world has virions. Measurements, as opposed to estimates, produce numbers almost as arresting. A litre of seawater may contain more than 100bn virions; a kilogram of dried soil perhaps a trillion.
Metagenomics, a part of biology that looks at all the nucleic acid in a given sample to get a sense of the range of life forms within it, reveals that these tiny throngs are highly diverse. A metagenomic analysis of two surveys of ocean life, the Tara Oceans and Malaspina missions, by Ahmed Zayed of Ohio State University, found evidence of 200,000 different species of virus. These diverse species play an enormous role in the ecology of the oceans.
A litre of seawater may contain 100bn virions; a kilogram of dried soil perhaps a trillion
On land, most of the photosynthesis which provides the biomass and energy needed for life takes place in plants. In the oceans, it is overwhelmingly the business of various sorts of bacteria and algae collectively known as phytoplankton. These creatures reproduce at a terrific rate, and viruses kill them at a terrific rate, too. According to work by Curtis Suttle of the University of British Columbia, bacterial phytoplankton typically last less than a week before being killed by viruses.
This increases the overall productivity of the oceans by helping bacteria recycle organic matter (it is easier for one cell to use the contents of another if a virus helpfully lets them free). It also goes some way towards explaining what the great mid-20th-century ecologist G. Evelyn Hutchinson called “the paradox of the plankton”. Given the limited nature of the resources that single-celled plankton need, you would expect a few species particularly well adapted to their use to dominate the ecosystem. Instead, the plankton display great variety. This may well be because whenever a particular form of plankton becomes dominant, its viruses expand with it, gnawing away at its comparative success.
It is also possible that this endless dance of death between viruses and microbes sets the stage for one of evolution’s great leaps forward. Many forms of single-celled plankton have molecular mechanisms that allow them to kill themselves. They are presumably used when one cell’s sacrifice allows its sister cells—which are genetically identical—to survive. One circumstance in which such sacrifice seems to make sense is when a cell is attacked by a virus. If the infected cell can kill itself quickly (a process called apoptosis) it can limit the number of virions the virus is able to make. This lessens the chances that other related cells nearby will die. Some bacteria have been shown to use this strategy; many other microbes are suspected of it.
There is another situation where self-sacrifice is becoming conduct for a cell: when it is part of a multicellular organism. As such organisms grow, cells that were once useful to them become redundant; they have to be got rid of. Eugene Koonin of America’s National Institutes of Health and his colleagues have explored the idea that virus-thwarting self-sacrifice and complexity-permitting self-sacrifice may be related, with the latter descended from the former. Dr Koonin’s model also suggests that the closer the cells are clustered together, the more likely this act of self-sacrifice is to have beneficial consequences.
For such profound propinquity, move from the free-flowing oceans to the more structured world of soil, where potential self-sacrificers can nestle next to each other. Its structure makes soil harder to sift for genes than water is. But last year Mary Firestone of the University of California, Berkeley, and her colleagues used metagenomics to count 3,884 new viral species in a patch of Californian grassland. That is undoubtedly an underestimate of the total diversity; their technique could see only viruses with RNA genomes, thus missing, among other things, most bacteriophages.
Metagenomics can also be applied to biological samples, such as bat guano in which it picks up viruses from both the bats and their food. But for the most part the finding of animal viruses requires more specific sampling. Over the course of the 2010s PREDICT, an American-government project aimed at finding animal viruses, gathered over 160,000 animal and human tissue samples from 35 countries and discovered 949 novel viruses.
The people who put together PREDICT now have grander plans. They want a Global Virome Project to track down all the viruses native to the world’s 7,400 species of mammals and waterfowl—the reservoirs most likely to harbour viruses capable of making the leap into human beings. In accordance with the more-predator-species-than-prey rule they expect such an effort would find about 1.5m viruses, of which around 700,000 might be able to infect humans. A planning meeting in 2018 suggested that such an undertaking might take ten years and cost $4bn. It looked like a lot of money then. Today those arguing for a system that can provide advance warning of the next pandemic make it sound pretty cheap.
IV Leaving their mark
THE TOLL which viruses have exacted throughout history suggests that they have left their mark on the human genome: things that kill people off in large numbers are powerful agents of natural selection. In 2016 David Enard, then at Stanford University and now at the University of Arizona, made a stab at showing just how much of the genome had been thus affected.
He and his colleagues started by identifying almost 10,000 proteins that seemed to be produced in all the mammals that had had their genomes sequenced up to that point. They then made a painstaking search of the scientific literature looking for proteins that had been shown to interact with viruses in some way or other. About 1,300 of the 10,000 turned up. About one in five of these proteins was connected to the immune system, and thus could be seen as having a professional interest in viral interaction. The others appeared to be proteins which the virus made use of in its attack on the host. The two cell-surface proteins that SARS–CoV-2 uses to make contact with its target cells and inveigle its way into them would fit into this category.
The researchers then compared the human versions of the genes for their 10,000 proteins with those in other mammals, and applied a statistical technique that distinguishes changes that have no real impact from the sort of changes which natural selection finds helpful and thus tries to keep. Genes for virus-associated proteins turned out to be evolutionary hotspots: 30% of all the adaptive change was seen in the genes for the 13% of the proteins which interacted with viruses. As quickly as viruses learn to recognise and subvert such proteins, hosts must learn to modify them.
A couple of years later, working with Dmitri Petrov at Stanford, Dr Enard showed that modern humans have borrowed some of these evolutionary responses to viruses from their nearest relatives. Around 2-3% of the DNA in an average European genome has Neanderthal origins, a result of interbreeding 50,000 to 30,000 years ago. For these genes to have persisted they must be doing something useful—otherwise natural selection would have removed them. Dr Enard and Dr Petrov found that a disproportionate number described virus-interacting proteins; of the bequests humans received from their now vanished relatives, ways to stay ahead of viruses seem to have been among the most important.
Viruses do not just shape the human genome through natural selection, though. They also insert themselves into it. At least a twelfth of the DNA in the human genome is derived from viruses; by some measures the total could be as high as a quarter.
Retroviruses like HIV are called retro because they do things backwards. Where cellular organisms make their RNA from DNA templates, retroviruses do the reverse, making DNA copies of their RNA genomes. The host cell obligingly makes these copies into double-stranded DNA which can be stitched into its own genome. If this happens in a cell destined to give rise to eggs or sperm, the viral genes are passed from parent to offspring, and on down the generations. Such integrated viral sequences, known as endogenous retroviruses (ERVs), account for 8% of the human genome.
This is another example of the way the same viral trick can be discovered a number of times. Many bacteriophages are also able to stitch copies of their genome into their host’s DNA, staying dormant, or “temperate”, for generations. If the cell is doing well and reproducing regularly, this quiescence is a good way for the viral genes to make more copies of themselves. When a virus senses that its easy ride may be coming to an end, though—for example, if the cell it is in shows signs of stress—it will abandon ship. What was latent becomes “lytic” as the viral genes produce a sufficient number of virions to tear the host apart.
Though some of their genes are associated with cancers, in humans ERVs do not burst back into action in later generations. Instead they have proved useful resources of genetic novelty. In the most celebrated example, at least ten different mammalian lineages make use of a retroviral gene for one of their most distinctively mammalian activities: building a placenta.
The placenta is a unique organ because it requires cells from the mother and the fetus to work together in order to pass oxygen and sustenance in one direction and carbon dioxide and waste in the other. One way this intimacy is achieved safely is through the creation of a tissue in which the membranes between cells are broken down to form a continuous sheet of cellular material.
The protein that allows new cells to merge themselves with this layer, syncytin-1, was originally used by retroviruses to join the external membranes of their virions to the external membranes of cells, thus gaining entry for the viral proteins and nucleic acids. Not only have different sorts of mammals co-opted this membrane-merging trick—other creatures have made use of it, too. The mabuya, a long-tailed skink which unusually for a lizard nurtures its young within its body, employs a retroviral syncytin protein to produce a mammalian-looking placenta. The most recent shared ancestor of mabuyas and mammals died out 80m years before the first dinosaur saw the light of day, but both have found the same way to make use of the viral gene.
You put your line-1 in, you take your line-1 out
This is not the only way that animals make use of their ERVs. Evidence has begun to accumulate that genetic sequences derived from ERVs are quite frequently used to regulate the activity of genes of more conventional origin. In particular, RNA molecules transcribed from an ERV called HERV-K play a crucial role in providing the stem cells found in embryos with their “pluripotency”—the ability to create specialised daughter cells of various different types. Unfortunately, when expressed in adults HERV-K can also be responsible for cancers of the testes.
As well as containing lots of semi-decrepit retroviruses that can be stripped for parts, the human genome also holds a great many copies of a “retrotransposon” called LINE-1. This a piece of DNA with a surprisingly virus-like way of life; it is thought by some biologists to have, like ERVs, a viral origin. In its full form, LINE-1 is a 6,000-letter sequence of DNA which describes a “reverse transcriptase” of the sort that retroviruses use to make DNA from their RNA genomes. When LINE-1 is transcribed into an mRNA and that mRNA subsequently translated to make proteins, the reverse transcriptase thus created immediately sets to work on the mRNA used to create it, using it as the template for a new piece of DNA which is then inserted back into the genome. That new piece of DNA is in principle identical to the piece that acted as the mRNA’s original template. The LINE-1 element has made a copy of itself.
In the 100m years or so that this has been going on in humans and the species from which they are descended the LINE-1 element has managed to pepper the genome with a staggering 500,000 copies of itself. All told, 17% of the human genome is taken up by these copies—twice as much as by the ERVs.
Most of the copies are severely truncated and incapable of copying themselves further. But some still have the knack, and this capability may be being put to good use. Fred Gage and his colleagues at the Salk Institute for Biological Studies, in San Diego, argue that LINE-1 elements have an important role in the development of the brain. In 2005 Dr Gage discovered that in mouse embryos—specifically, in the brains of those embryos—about 3,000 LINE-1 elements are still able to operate as retrotransposons, putting new copies of themselves into the genome of a cell and thus of all its descendants.
Brains develop through proliferation followed by pruning. First, nerve cells multiply pell-mell; then the cell-suicide process that makes complex life possible prunes them back in a way that looks a lot like natural selection. Dr Gage suspects that the movement of LINE-1 transposons provides the variety in the cell population needed for this selection process. Choosing between cells with LINE-1 in different places, he thinks, could be a key part of the process from which the eventual neural architecture emerges. What is true in mice is, as he showed in 2009, true in humans, too. He is currently developing a technique for looking at the process in detail by comparing, post mortem, the genomes of different brain cells from single individuals to see if their LINE-1 patterns vary in the ways that his theory would predict.
V Promised lands
HUMAN EVOLUTION may have used viral genes to make big-brained live-born life possible; but viral evolution has used them to kill off those big brains on a scale that is easily forgotten. Compare the toll to that of war. In the 20th century, the bloodiest in human history, somewhere between 100m and 200m people died as a result of warfare. The number killed by measles was somewhere in the same range; the number who died of influenza probably towards the top of it; and the number killed by smallpox—300m-500m—well beyond it. That is why the eradication of smallpox from the wild, achieved in 1979 by a globally co-ordinated set of vaccination campaigns, stands as one of the all-time-great humanitarian triumphs.
Other eradications should eventually follow. Even in their absence, vaccination has led to a steep decline in viral deaths. But viruses against which there is no vaccine, either because they are very new, like SARS–CoV-2, or peculiarly sneaky, like HIV, can still kill millions.
Reducing those tolls is a vital aim both for research and for public-health policy. Understandably, a far lower priority is put on the benefits that viruses can bring. This is mostly because they are as yet much less dramatic. They are also much less well understood.
The viruses most prevalent in the human body are not those which infect human cells. They are those which infect the bacteria that live on the body’s surfaces, internal and external. The average human “microbiome” harbours perhaps 100trn of these bacteria. And where there are bacteria, there are bacteriophages shaping their population.
The microbiome is vital for good health; when it goes wrong it can mess up a lot else. Gut bacteria seem to have a role in maintaining, and possibly also causing, obesity in the well-fed and, conversely, in tipping the poorly fed into a form of malnutrition called kwashiorkor. Ill-regulated gut bacteria have also been linked, if not always conclusively, with diabetes, heart disease, cancers, depression and autism. In light of all this, the question “who guards the bacterial guardians?” is starting to be asked.
The viruses that prey on the bacteria are an obvious answer. Because the health of their host’s host—the possessor of the gut they find themselves in—matters to these phages, they have an interest in keeping the microbiome balanced. Unbalanced microbiomes allow pathogens to get a foothold. This may explain a curious detail of a therapy now being used as a treatment of last resort against Clostridium difficile, a bacterium that causes life-threatening dysentery. The therapy in question uses a transfusion of faecal matter, with its attendant microbes, from a healthy individual to reboot the patient’s microbiome. Such transplants, it appears, are more likely to succeed if their phage population is particularly diverse.
Medicine is a very long way from being able to use phages to fine-tune the microbiome. But if a way of doing so is found, it will not in itself be a revolution. Attempts to use phages to promote human health go back to their discovery in 1917, by Félix d’Hérelle, a French microbiologist, though those early attempts at therapy were not looking to restore balance and harmony. On the basis that the enemy of my enemy is my friend, doctors simply treated bacterial infections with phages thought likely to kill the bacteria.
The arrival of antibiotics saw phage therapy abandoned in most places, though it persisted in the Soviet Union and its satellites. Various biotechnology companies think they may now be able to revive the tradition—and make it more effective. One option is to remove the bits of the viral genome that let phages settle down to a temperate life in a bacterial genome, leaving them no option but to keep on killing. Another is to write their genes in ways that avoid the defences with which bacteria slice up foreign DNA.
The hope is that phage therapy will become a backup in difficult cases, such as infection with antibiotic-resistant bugs. There have been a couple of well-publicised one-off successes outside phage therapy’s post-Soviet homelands. In 2016 Tom Patterson, a researcher at the University of California, San Diego, was successfully treated for an antibiotic-resistant bacterial infection with specially selected (but un-engineered) phages. In 2018 Graham Hatfull of the University of Pittsburgh used a mixture of phages, some engineered so as to be incapable of temperance, to treat a 16-year-old British girl who had a bad bacterial infection after a lung transplant. Clinical trials are now getting under way for phage treatments aimed at urinary-tract infections caused by Escherichia coli, Staphylococcus aureus infections that can lead to sepsis and Pseudomonas aeruginosa infections that cause complications in people who have cystic fibrosis.
Viruses which attack bacteria are not the only ones genetic engineers have their eyes on. Engineered viruses are of increasing interest to vaccine-makers, to cancer researchers and to those who want to treat diseases by either adding new genes to the genome or disabling faulty ones. If you want to get a gene into a specific type of cell, a virion that recognises something about such cells may often prove a good tool.
The vaccine used to contain the Ebola outbreak in the Democratic Republic of Congo over the past two years was made by engineering Indiana vesiculovirus, which infects humans but cannot reproduce in them, so that it expresses a protein found on the surface of the Ebola virus; thus primed, the immune system responds to Ebola much more effectively. The World Health Organisation’s current list of 29 covid-19 vaccines in clinical trials features six versions of other viruses engineered to look a bit like SARS-CoV-2. One is based on a strain of measles that has long been used as a vaccine against that disease.
Viruses engineered to engender immunity against pathogens, to kill cancer cells or to encourage the immune system to attack them, or to deliver needed genes to faulty cells all seem likely to find their way into health care. Other engineered viruses are more worrying. One way to understand how viruses spread and kill is to try and make particularly virulent ones. In 2005, for example, Terrence Tumpey of America’s Centres for Disease Control and Prevention and his colleagues tried to understand the deadliness of the influenza virus responsible for the pandemic of 1918-20 by taking a more benign strain, adding what seemed to be distinctive about the deadlier one and trying out the result on mice. It was every bit as deadly as the original, wholly natural version had been.
The use of engineered pathogens as weapons of war is of dubious utility, completely illegal and repugnant to almost all
Because such “gain of function” research could, if ill-conceived or poorly implemented, do terrible damage, it requires careful monitoring. And although the use of engineered pathogens as weapons of war is of dubious utility—such weapons are hard to aim and hard to stand down, and it is not easy to know how much damage they have done—as well as being completely illegal and repugnant to almost all, such possibilities will and should remain a matter of global concern.
Information which, for billions of years, has only ever come into its own within infected cells can now be inspected on computer screens and rewritten at will. The power that brings is sobering. It marks a change in the history of both viruses and people—a change which is perhaps as important as any of those made by modern biology. It is constraining a small part of the viral world in a way which, so far, has been to people’s benefit. It is revealing that world’s further reaches in a way which cannot but engender awe. ■
Editor’s note: Some of our covid-19 coverage is free for readers of The Economist Today, our daily newsletter. For more stories and our pandemic tracker, see our hub
This article appeared in the Essay section of the print edition under the headline “The outsiders inside”
HUMANS THINK of themselves as the world’s apex predators. Hence the silence of sabre-tooth tigers, the absence of moas from New Zealand and the long list of endangered megafauna. But SARS–CoV-2 shows how people can also end up as prey. Viruses have caused a litany of modern pandemics, from covid-19, to HIV/AIDS to the influenza outbreak in 1918-20, which killed many more people than the first world war. Before that, the colonisation of the Americas by Europeans was abetted—and perhaps made possible—by epidemics of smallpox, measles and influenza brought unwittingly by the invaders, which annihilated many of the original inhabitants.
The influence of viruses on life on Earth, though, goes far beyond the past and present tragedies of a single species, however pressing they seem. Though the study of viruses began as an investigation into what appeared to be a strange subset of pathogens, recent research puts them at the heart of an explanation of the strategies of genes, both selfish and otherwise.
Viruses are unimaginably varied and ubiquitous. And it is becoming clear just how much they have shaped the evolution of all organisms since the very beginnings of life. In this, they demonstrate the blind, pitiless power of natural selection at its most dramatic. And—for one group of brainy bipedal mammals that viruses helped create—they also present a heady mix of threat and opportunity.
As our essay in this week’s issue explains, viruses are best thought of as packages of genetic material that exploit another organism’s metabolism in order to reproduce. They are parasites of the purest kind: they borrow everything from the host except the genetic code that makes them what they are. They strip down life itself to the bare essentials of information and its replication. If the abundance of viruses is anything to go by, that is a very successful strategy indeed.
The world is teeming with them. One analysis of seawater found 200,000 different viral species, and it was not setting out to be comprehensive. Other research suggests that a single litre of seawater may contain more than 100bn virus particles, and a kilo of dried soil ten times that number. Altogether, according to calculations on the back of a very big envelope, the world might contain 1031 of the things—that is one followed by 31 zeros, far outnumbering all other forms of life on the planet.
As far as anyone can tell, viruses—often of many different sorts—have adapted to attack every organism that exists. One reason they are powerhouses of evolution is that they oversee a relentless and prodigious slaughter, mutating as they do so. This is particularly clear in the oceans, where a fifth of single-celled plankton are killed by viruses every day. Ecologically, this promotes diversity by scything down abundant species, thus making room for rarer ones. The more common an organism, the more likely it is that a local plague of viruses specialised to attack it will develop, and so keep it in check.
This propensity to cause plagues is also a powerful evolutionary stimulus for prey to develop defences, and these defences sometimes have wider consequences. For example, one explanation for why a cell may deliberately destroy itself is if its sacrifice lowers the viral load on closely related cells nearby. That way, its genes, copied in neighbouring cells, are more likely to survive. It so happens that such altruistic suicide is a prerequisite for cells to come together and form complex organisms, such as pea plants, mushrooms and human beings.
The other reason viruses are engines of evolution is that they are transport mechanisms for genetic information. Some viral genomes end up integrated into the cells of their hosts, where they can be passed down to those organisms’ descendants. Between 8% and 25% of the human genome seems to have such viral origins. But the viruses themselves can in turn be hijacked, and their genes turned to new uses. For example, the ability of mammals to bear live young is a consequence of a viral gene being modified to permit the formation of placentas. And even human brains may owe their development in part to the movement within them of virus-like elements that create genetic differences between neurons within a single organism.
Evolution’s most enthralling insight is that breathtaking complexity can emerge from the sustained, implacable and nihilistic competition within and between organisms. The fact that the blind watchmaker has equipped you with the capacity to read and understand these words is in part a response to the actions of swarms of tiny, attacking replicators that have been going on, probably, since life first emerged on Earth around 4bn years ago. It is a startling example of that principle in action—and viruses have not finished yet.
Humanity’s unique, virus-chiselled consciousness opens up new avenues to deal with the viral threat and to exploit it. This starts with the miracle of vaccination, which defends against a pathogenic attack before it is launched. Thanks to vaccines, smallpox is no more, having taken some 300m lives in the 20th century. Polio will one day surely follow. New research prompted by the covid-19 pandemic will enhance the power to examine the viral realm and the best responses to it that bodies can muster—taking the defence against viruses to a new level.
Another avenue for progress lies in the tools for manipulating organisms that will come from an understanding of viruses and the defences against them. Early versions of genetic engineering relied on restriction enzymes—molecular scissors with which bacteria cut up viral genes and which biotechnologists employ to move genes around. The latest iteration of biotechnology, gene editing letter by letter, which is known as CRISPR, makes use of a more precise antiviral mechanism.
From the smallest beginnings
The natural world is not kind. A virus-free existence is an impossibility so deeply unachievable that its desirability is meaningless. In any case, the marvellous diversity of life rests on viruses which, as much as they are a source of death, are also a source of richness and of change. Marvellous, too, is the prospect of a world where viruses become a source of new understanding for humans—and kill fewer of them than ever before. ■
Correction: An earlier version of this article got its maths wrong. 1031 is one followed by 31 zeroes, not ten followed by 31 zeroes as we first wrote. Sorry.
In 1942, the anthropologist Ashley Montagu published “Man’s Most Dangerous Myth: The Fallacy of Race,” an influential book that argued that race is a social concept with no genetic basis. A classic example often cited is the inconsistent definition of “black.” In the United States, historically, a person is “black” if he has any sub-Saharan African ancestry; in Brazil, a person is not “black” if he is known to have any European ancestry. If “black” refers to different people in different contexts, how can there be any genetic basis to it?
Beginning in 1972, genetic findings began to be incorporated into this argument. That year, the geneticist Richard Lewontin published an important study of variation in protein types in blood. He grouped the human populations he analyzed into seven “races” — West Eurasians, Africans, East Asians, South Asians, Native Americans, Oceanians and Australians — and found that around 85 percent of variation in the protein types could be accounted for by variation within populations and “races,” and only 15 percent by variation across them. To the extent that there was variation among humans, he concluded, most of it was because of “differences between individuals.”
In this way, a consensus was established that among human populations there are no differences large enough to support the concept of “biological race.” Instead, it was argued, race is a “social construct,” a way of categorizing people that changes over time and across countries.
It is true that race is a social construct. It is also true, as Dr. Lewontin wrote, that human populations “are remarkably similar to each other” from a genetic point of view.
But over the years this consensus has morphed, seemingly without questioning, into an orthodoxy. The orthodoxy maintains that the average genetic differences among people grouped according to today’s racial terms are so trivial when it comes to any meaningful biological traits that those differences can be ignored.
The orthodoxy goes further, holding that we should be anxious about any research into genetic differences among populations. The concern is that such research, no matter how well-intentioned, is located on a slippery slope that leads to the kinds of pseudoscientific arguments about biological difference that were used in the past to try to justify the slave trade, the eugenics movement and the Nazis’ murder of six million Jews.
I have deep sympathy for the concern that genetic discoveries could be misused to justify racism. But as a geneticist I also know that it is simply no longer possible to ignore average genetic differences among “races.”
Groundbreaking advances in DNA sequencing technology have been made over the last two decades. These advances enable us to measure with exquisite accuracy what fraction of an individual’s genetic ancestry traces back to, say, West Africa 500 years ago — before the mixing in the Americas of the West African and European gene pools that were almost completely isolated for the last 70,000 years. With the help of these tools, we are learning that while race may be a social construct, differences in genetic ancestry that happen to correlate to many of today’s racial constructs are real.
Recent genetic studies have demonstrated differences across populations not just in the genetic determinants of simple traits such as skin color, but also in more complex traits like bodily dimensions and susceptibility to diseases. For example, we now know that genetic factors help explain why northern Europeans are taller on average than southern Europeans, why multiple sclerosis is more common in European-Americans than in African-Americans, and why the reverse is true for end-stage kidney disease.
I am worried that well-meaning people who deny the possibility of substantial biological differences among human populations are digging themselves into an indefensible position, one that will not survive the onslaught of science. I am also worried that whatever discoveries are made — and we truly have no idea yet what they will be — will be cited as “scientific proof” that racist prejudices and agendas have been correct all along, and that those well-meaning people will not understand the science well enough to push back against these claims.
This is why it is important, even urgent, that we develop a candid and scientifically up-to-date way of discussing any such differences, instead of sticking our heads in the sand and being caught unprepared when they are found.
To get a sense of what modern genetic research into average biological differences across populations looks like, consider an example from my own work. Beginning around 2003, I began exploring whether the population mixture that has occurred in the last few hundred years in the Americas could be leveraged to find risk factors for prostate cancer, a disease that occurs 1.7 times more often in self-identified African-Americans than in self-identified European-Americans. This disparity had not been possible to explain based on dietary and environmental differences, suggesting that genetic factors might play a role.
Self-identified African-Americans turn out to derive, on average, about 80 percent of their genetic ancestry from enslaved Africans brought to America between the 16th and 19th centuries. My colleagues and I searched, in 1,597 African-American men with prostate cancer, for locations in the genome where the fraction of genes contributed by West African ancestors was larger than it was elsewhere in the genome. In 2006, we found exactly what we were looking for: a location in the genome with about 2.8 percent more African ancestry than the average.
When we looked in more detail, we found that this region contained at least seven independent risk factors for prostate cancer, all more common in West Africans. Our findings could fully account for the higher rate of prostate cancer in African-Americans than in European-Americans. We could conclude this because African-Americans who happen to have entirely European ancestry in this small section of their genomes had about the same risk for prostate cancer as random Europeans.
Did this research rely on terms like “African-American” and “European-American” that are socially constructed, and did it label segments of the genome as being probably “West African” or “European” in origin? Yes. Did this research identify real risk factors for disease that differ in frequency across those populations, leading to discoveries with the potential to improve health and save lives? Yes.
While most people will agree that finding a genetic explanation for an elevated rate of disease is important, they often draw the line there. Finding genetic influences on a propensity for disease is one thing, they argue, but looking for such influences on behavior and cognition is another.
But whether we like it or not, that line has already been crossed. A recent study led by the economist Daniel Benjamin compiled information on the number of years of education from more than 400,000 people, almost all of whom were of European ancestry. After controlling for differences in socioeconomic background, he and his colleagues identified 74 genetic variations that are over-represented in genes known to be important in neurological development, each of which is incontrovertibly more common in Europeans with more years of education than in Europeans with fewer years of education.
It is not yet clear how these genetic variations operate. A follow-up study of Icelanders led by the geneticist Augustine Kong showed that these genetic variations also nudge people who carry them to delay having children. So these variations may be explaining longer times at school by affecting a behavior that has nothing to do with intelligence.
This study has been joined by others finding genetic predictors of behavior. One of these, led by the geneticist Danielle Posthuma, studied more than 70,000 people and found genetic variations in more than 20 genes that were predictive of performance on intelligence tests.
Is performance on an intelligence test or the number of years of school a person attends shaped by the way a person is brought up? Of course. But does it measure something having to do with some aspect of behavior or cognition? Almost certainly. And since all traits influenced by genetics are expected to differ across populations (because the frequencies of genetic variations are rarely exactly the same across populations), the genetic influences on behavior and cognition will differ across populations, too.
You will sometimes hear that any biological differences among populations are likely to be small, because humans have diverged too recently from common ancestors for substantial differences to have arisen under the pressure of natural selection. This is not true. The ancestors of East Asians, Europeans, West Africans and Australians were, until recently, almost completely isolated from one another for 40,000 years or longer, which is more than sufficient time for the forces of evolution to work. Indeed, the study led by Dr. Kong showed that in Iceland, there has been measurable genetic selection against the genetic variations that predict more years of education in that population just within the last century.
To understand why it is so dangerous for geneticists and anthropologists to simply repeat the old consensus about human population differences, consider what kinds of voices are filling the void that our silence is creating. Nicholas Wade, a longtime science journalist for The New York Times, rightly notes in his 2014 book, “A Troublesome Inheritance: Genes, Race and Human History,” that modern research is challenging our thinking about the nature of human population differences. But he goes on to make the unfounded and irresponsible claim that this research is suggesting that genetic factors explain traditional stereotypes.
One of Mr. Wade’s key sources, for example, is the anthropologist Henry Harpending, who has asserted that people of sub-Saharan African ancestry have no propensity to work when they don’t have to because, he claims, they did not go through the type of natural selection for hard work in the last thousands of years that some Eurasians did. There is simply no scientific evidence to support this statement. Indeed, as 139 geneticists (including myself) pointed out in a letter to The New York Times about Mr. Wade’s book, there is no genetic evidence to back up any of the racist stereotypes he promotes.
Another high-profile example is James Watson, the scientist who in 1953 co-discovered the structure of DNA, and who was forced to retire as head of the Cold Spring Harbor Laboratories in 2007 after he stated in an interview — without any scientific evidence — that research has suggested that genetic factors contribute to lower intelligence in Africans than in Europeans.
At a meeting a few years later, Dr. Watson said to me and my fellow geneticist Beth Shapiro something to the effect of “When are you guys going to figure out why it is that you Jews are so much smarter than everyone else?” He asserted that Jews were high achievers because of genetic advantages conferred by thousands of years of natural selection to be scholars, and that East Asian students tended to be conformist because of selection for conformity in ancient Chinese society. (Contacted recently, Dr. Watson denied having made these statements, maintaining that they do not represent his views; Dr. Shapiro said that her recollection matched mine.)
What makes Dr. Watson’s and Mr. Wade’s statements so insidious is that they start with the accurate observation that many academics are implausibly denying the possibility of average genetic differences among human populations, and then end with a claim — backed by no evidence — that they know what those differences are and that they correspond to racist stereotypes. They use the reluctance of the academic community to openly discuss these fraught issues to provide rhetorical cover for hateful ideas and old racist canards.
This is why knowledgeable scientists must speak out. If we abstain from laying out a rational framework for discussing differences among populations, we risk losing the trust of the public and we actively contribute to the distrust of expertise that is now so prevalent. We leave a vacuum that gets filled by pseudoscience, an outcome that is far worse than anything we could achieve by talking openly.
If scientists can be confident of anything, it is that whatever we currently believe about the genetic nature of differences among populations is most likely wrong. For example, my laboratory discovered in 2016, based on our sequencing of ancient human genomes, that “whites” are not derived from a population that existed from time immemorial, as some people believe. Instead, “whites” represent a mixture of four ancient populations that lived 10,000 years ago and were each as different from one another as Europeans and East Asians are today.
So how should we prepare for the likelihood that in the coming years, genetic studies will show that many traits are influenced by genetic variations, and that these traits will differ on average across human populations? It will be impossible — indeed, anti-scientific, foolish and absurd — to deny those differences.
For me, a natural response to the challenge is to learn from the example of the biological differences that exist between males and females. The differences between the sexes are far more profound than those that exist among human populations, reflecting more than 100 million years of evolution and adaptation. Males and females differ by huge tracts of genetic material — a Y chromosome that males have and that females don’t, and a second X chromosome that females have and males don’t.
Most everyone accepts that the biological differences between males and females are profound. In addition to anatomical differences, men and women exhibit average differences in size and physical strength. (There are also average differences in temperament and behavior, though there are important unresolved questions about the extent to which these differences are influenced by social expectations and upbringing.)
How do we accommodate the biological differences between men and women? I think the answer is obvious: We should both recognize that genetic differences between males and females exist and we should accord each sex the same freedoms and opportunities regardless of those differences.
It is clear from the inequities that persist between women and men in our society that fulfilling these aspirations in practice is a challenge. Yet conceptually it is straightforward. And if this is the case with men and women, then it is surely the case with whatever differences we may find among human populations, the great majority of which will be far less profound.
An abiding challenge for our civilization is to treat each human being as an individual and to empower all people, regardless of what hand they are dealt from the deck of life. Compared with the enormous differences that exist among individuals, differences among populations are on average many times smaller, so it should be only a modest challenge to accommodate a reality in which the average genetic contributions to human traits differ.
It is important to face whatever science will reveal without prejudging the outcome and with the confidence that we can be mature enough to handle any findings. Arguing that no substantial differences among human populations are possible will only invite the racist misuse of genetics that we wish to avoid.
David Reich is a professor of genetics at Harvard and the author of the forthcoming book “Who We Are and How We Got Here: Ancient DNA and the New Science of the Human Past,” from which this article is adapted.
Vanderbilt biologist Nicole Creanza Nicole Creanza takes interdisciplinary approach to human evolution as guest editor of Royal Society journal
The evolution of human biology should be considered part and parcel with the evolution of humanity itself, proposes Nicole Creanza, assistant professor of biological sciences. She is the guest editor of a new themed issue of the Philosophical Transactions of the Royal Society B, the oldest scientific journal in the world, that focuses on an interdisciplinary approach to human evolution.
Stanford professor Marc Feldman and Stanford postdoc Oren Kolodny collaborated with Creanza on the special issue.
“Within the blink of an eye on a geological timescale, humans advanced from using basic stone tools to examining the rocks on Mars; however, our exact evolutionary path and the relative importance of genetic and cultural evolution remain a mystery,” said Creanza, who specializes in the application of computational and theoretical approaches to human and cultural evolution, particularly language development. “Our cultural capacities-to create new ideas, to communicate and learn from one another, and to form vast social networks-together make us uniquely human, but the origins, the mechanisms, and the evolutionary impact of these capacities remain unknown.”
The special issue brings together researchers in biology, anthropology, archaeology, economics, psychology, computer science and more to explore the cultural forces affecting human evolution from a wider perspective than is usually taken.
“Researchers have begun to recognize that understanding non-genetic inheritance, including culture, ecology, the microbiome, and regulation of gene expression, is fundamental to fully comprehending evolution,” said Creanza. “It is essential to understand the dynamics of cultural inheritance at different temporal and spatial scales, to uncover the underlying mechanisms that drive these dynamics, and to shed light on their implications for our current theory of evolution as well as for our interpretation and predictions regarding human behavior.”
In addition to an essay discussing the need for an interdisciplinary approach to human evolution, Creanza included an interdisciplinary study of her own, examining the origins of English’s contribution to Sranan, a creole that emerged in Suriname following an influx of indentured servants from England in the 17th century.
Creanza, along with linguists Andre Sherriah and Hubert Devonish of the University of the West Indes and psychologist Ewart Thomas from Stanford, sought to determine the geographic origins of the English speakers whose regional dialects formed the backbone of Sranan. Their work combined linguistic, historical and genetic approaches to determine that the English speakers who influenced Sranan the most originated largely from two counties on opposite sides of southern England: Bristol, in the west, and Essex, in the east.
“Thus, analyzing the features of modern-day languages might give us new information about events in human history that left few other traces,” Creanza said.
Three different studies, done by different teams of scientists proved something really extraordinary. But when a new research connected these 3 discoveries, something shocking was realized, something hiding in plain sight.
Human emotion literally shapes the world around us. Not just our perception of the world, but reality itself.
In the first experiment, human DNA, isolated in a sealed container, was placed near a test subject. Scientists gave the donor emotional stimulus and fascinatingly enough, the emotions affected their DNA in the other room.
In the presence of negative emotions the DNA tightened. In the presence of positive emotions the coils of the DNA relaxed.
The scientists concluded that “Human emotion produces effects which defy conventional laws of physics.”
In the second, similar but unrelated experiment, different group of scientists extracted Leukocytes (white blood cells) from donors and placed into chambers so they could measure electrical changes.
In this experiment, the donor was placed in one room and subjected to “emotional stimulation” consisting of video clips, which generated different emotions in the donor.
The DNA was placed in a different room in the same building. Both the donor and his DNA were monitored and as the donor exhibited emotional peaks or valleys (measured by electrical responses), the DNA exhibited the IDENTICAL RESPONSES AT THE EXACT SAME TIME.
There was no lag time, no transmission time. The DNA peaks and valleys EXACTLY MATCHED the peaks and valleys of the donor in time.
The scientists wanted to see how far away they could separate the donor from his DNA and still get this effect. They stopped testing after they separated the DNA and the donor by 50 miles and STILL had the SAME result. No lag time; no transmission time.
The DNA and the donor had the same identical responses in time. The conclusion was that the donor and the DNA can communicate beyond space and time.
The third experiment proved something pretty shocking!
Scientists observed the effect of DNA on our physical world.
Light photons, which make up the world around us, were observed inside a vacuum. Their natural locations were completely random.
Human DNA was then inserted into the vacuum. Shockingly the photons were no longer acting random. They precisely followed the geometry of the DNA.
Scientists who were studying this, described the photons behaving “surprisingly and counter-intuitively”. They went on to say that “We are forced to accept the possibility of some new field of energy!”
They concluded that human DNA literally shape the behavior of light photons that make up the world around us!
So when a new research was done, and all of these 3 scientific claims were connected together, scientists were shocked.
They came to a stunning realization that if our emotions affect our DNA and our DNA shapes the world around us, than our emotions physically change the world around us.
And not just that, we are connected to our DNA beyond space and time.
We create our reality by choosing it with our feelings.
Science has already proven some pretty MINDBLOWING facts about The Universe we live in. All we have to do is connect the dots.
Experimentos com ratos feitos por pesquisadores de universidades de São Paulo reforçam a ideia de que o excesso de peso pode ser um fenômeno que transcende gerações – e não apenas porque os filhos tendem a herdar dos pais genes que favorecem o acúmulo de energia e os tornam predispostos à obesidade ou porque vivem em um ambiente com disponibilidade excessiva de comida. Alterações na oferta de alimento para as fêmeas um pouco antes ou durante a gravidez parecem aumentar, por mecanismos ainda pouco compreendidos, a probabilidade de que tenham filhos e até netos com sobrepeso.
Em uma série de testes, a bióloga Maria Martha Bernardi e sua equipe na Universidade Paulista (Unip) alimentaram algumas ratas no início da vida reprodutiva e outras já grávidas com uma dieta bastante calórica e aguardaram para ver o que acontecia com a primeira geração de filhotes e também com os filhos desses filhotes. Tanto os roedores que nasceram de mães superalimentadas quanto os da geração seguinte apresentaram mais predisposição a desenvolver sobrepeso.
A tendência de ganho excessivo de peso ocorreu mesmo quando os filhos e os netos dessas ratas foram alimentados apenas com a dieta padrão de laboratório. Segundo Martha, esses resultados indicam que o período em que o feto está se desenvolvendo no útero é crucial para definir a regulação do metabolismo do animal e, ao menos, o da geração seguinte.
Se essas mudanças aparecessem apenas na primeira geração, o mais natural seria imaginar que alterações hormonais provocadas pela dieta materna teriam afetado os filhotes. Como o efeito avança até a segunda geração, os pesquisadores suspeitam que a propensão a ganhar peso seja mantida por mecanismos epigenéticos: alterações no padrão de ativação e desligamento dos genes provocadas por fatores ambientais, como a dieta, e transmitidas às gerações seguintes. Essas mudanças no perfil de acionamento dos genes não alteram diretamente a sequência de “letras químicas” do DNA, apesar de serem herdadas através das gerações. Embora o grupo de Martha não tenha analisado o padrão de atividade dos genes, dados obtidos por cientistas mundo afora indicam que mudanças no perfil de ativação gênica sem alteração na sequência de DNA podem acontecer tanto em animais quanto em seres humanos.
Dieta que engorda
Curiosamente, não foi só a superalimentação materna durante a gestação que parece ter mexido com o perfil de ativação de seus genes e deixado filhos e netos com tendência a engordar. Em um dos experimentos, realizado em parceria com pesquisadores das universidades de São Paulo (USP), Federal do ABC (UFABC) e Santo Amaro (Unisa), 12 fêmeas de ratos receberam 40% menos comida do que o considerado normal para as roedoras prenhes, enquanto oito ratas do grupo de controle foram alimentadas com a dieta habitual de laboratório.
As fêmeas que passaram fome durante a gestação ganharam menos da metade do peso das ratas que puderam comer à vontade. Os filhotes das mães submetidas à restrição alimentar nasceram menores e continuaram mais magros durante algum tempo, ainda que recebessem a mesma quantidade de comida que os filhos das ratas que não passaram fome. Só na idade adulta a diferença desapareceu e os dois grupos de roedores alcançaram peso semelhante, embora os filhos das ratas famintas apresentassem uma proporção maior de gordura corporal – em especial, de uma forma de gordura que se acumula entre os órgãos (gordura visceral), associada a maior risco de problemas cardiovasculares.
A diferença mais importante surgiu na segunda geração. Os netos de ratas que haviam comido pouco enquanto estavam prenhes nasceram menores, mas, depois de adultos, eram um pouco (de 10% a 15%) mais pesados que os netos das ratas alimentadas normalmente. Eles tinham mais gordura visceral e também sinais de inflamação no cérebro. Esse ganho extra de peso ocorreu mesmo com as fêmeas da primeira geração, portanto, mães desses animais, tendo sido alimentadas normalmente. É como se a privação de alimento experimentada pelas ratas da geração inicial provocasse uma reprogramação metabólica duradoura em seus descendentes, afirmam os pesquisadores em artigo publicado em maio de 2016 na revista Reproduction, Fertility and Development.
O trabalho da equipe paulista, nesse ponto, confirma pesquisas anteriores que já haviam encontrado uma associação entre episódios de fome na gravidez e o nascimento de filhos com propensão ao aumento de peso e aos problemas de saúde a ele associados. Embora não tenham identificado o mecanismo específico por trás desse efeito, Martha Bernardi e sua equipe suspeitam que compostos produzidos pelo organismo das mães da geração inicial, parcialmente privadas de comida na gestação, ativem genes que favorecem o rápido ganho de peso no filhote. Assim, os sinais químicos emitidos pelo corpo materno funcionariam como um alerta de que o ambiente é de escassez e que é preciso usar com máxima eficiência os recursos alimentares disponíveis. Essa sinalização recebida pelo organismo do filhote poderia fazer toda a diferença, representando a chance de crescer e sobreviver em um ambiente com privação de alimento. “Mas também pode levar à obesidade, caso a oferta de alimentos volte a se normalizar depois que ele nasce”, explica Martha.
Estudos realizados nas décadas anteriores mostraram uma situação muito parecida com a descrita acima entre os descendentes das mulheres que ficaram grávidas durante o chamado Hongerwinter (inverno da fome, em holandês), quando os exércitos nazistas que recuavam na Holanda diante do avanço dos Aliados cortaram boa parte do transporte de suprimentos para o país entre o fim de 1944 e o começo de 1945, no final da Segunda Grande Guerra. Tanto os filhos quanto os netos das sobreviventes do Hongerwinter apresentavam taxas de obesidade e problemas metabólicos acima do esperado para a população geral.
Inflamação no cérebro
Em outro estudo, Martha e seus colegas forneceram alimentação hipercalórica – uma mistura de ração padrão mais um suplemento líquido rico em diferentes tipos de gordura – para 10 ratas logo após o desmame, enquanto outro grupo de fêmeas recebeu a alimentação normal e serviu de controle. Conforme o esperado, as ratas submetidas à dieta hipercalórica quando bebês ficaram acima do peso, ainda que não obesas, ao chegar à puberdade. Efeitos semelhantes foram observados em suas filhas: eram ratas que, quando adultas, apresentaram sobrepeso e alterações metabólicas, como o acúmulo de gordura visceral, embora tenham sido tratadas apenas com uma dieta balanceada durante toda a vida. Também publicado na Reproduction, Fertility and Development, esse trabalho e outros estudos do grupo indicam que o sobrepeso foi o desencadeador de processos inflamatórios que afetaram o cérebro da mãe e da prole, de forma aparentemente duradoura.
Se parece estranho imaginar que o excesso de peso pode levar a uma inflamação cerebral, é preciso lembrar que as células de gordura não são meros depósitos de calorias. Os adipócitos, como são chamados, produzem uma grande variedade de substâncias, entre as quais moléculas desencadeadoras de inflamações, que chegam à corrente sanguínea e, a partir dela, ao hipotálamo, região do cérebro associada, entre outras funções, ao controle da fome.
Trabalhos do grupo da Unip ainda não publicados indicam ainda que essa inflamação pode atingir outras áreas cerebrais dos roedores. A hipótese dos pesquisadores é de que o processo inflamatório no órgão esteja ligado à reprogramação do organismo transmitida da mãe para os filhotes, incluindo aí alterações no controle do apetite que podem se manter durante a vida adulta.
Para Alicia Kowaltowski, pesquisadora do Instituto de Química da USP que estuda a relação entre a dieta e os mecanismos de produção de energia das células, é bastante forte a possibilidade de que a tendência ao sobrepeso e à obesidade seja passada de uma geração para outra por meios que não envolvem a herança de genes favorecedores do ganho de peso. “A questão é saber quais são os mecanismos que estão por trás desses fenômenos”, conta a pesquisadora.
Entre tais mecanismos, um candidato que tem ganhado força são as transformações epigenéticas. O prefixo grego epi significa superior, e na palavra epigenética, cunhada nos anos 1940 pelo embriologista inglês Conrad Waddington, designa a área da biologia que estuda as modificações químicas motivadas pelo ambiente que levam à ativação ou inativação dos genes e alteram o funcionamento do organismo. Uma das modificações químicas mais comuns e simples sofrida pelos genes é a chamada metilação. Nela, um grupo metila, formado por um átomo de carbono e três de hidrogênio (CH3), acopla-se a um trecho de DNA, impedindo que ele seja lido pelo maquinário da célula. O resultado é o silenciamento daquela região. Estudos com dezenas de espécies de animais, plantas e fungos já mostraram que o perfil de metilação pode ser transmitido de uma geração para outra e afetar as características da prole.
Influência paterna
O papel das mães no sobrepeso dos filhos parece cada vez mais sólido. E quanto ao papel do pai? “Há alguns indícios de que a influência paterna também pode ocorrer, mas eles são menos claros”, diz Martha Bernardi. Por um lado, faz sentido que influências epigenéticas possam ser transmitidas pelo lado paterno – assim como outras células do organismo, os espermatozoides podem ser afetados por alterações no padrão de ativação dos genes produzidas por influência do ambiente. Se tais mudanças não forem totalmente eliminadas após o encontro entre as células sexuais masculinas e os óvulos, o novo indivíduo gerado pela fecundação poderia carregar parte da memória epigenética de seu pai.
Um estudo de 2015, feito por uma equipe da Universidade de Copenhague, na Dinamarca, e liderado por Romain Barrès, mostrou que esse cenário é plausível ao estudar os espermatozoides de 16 homens obesos e outros 10 com peso normal. No caso dos voluntários obesos, os padrões epigenéticos, como os de metilação, concentravam-se em genes ligados ao desenvolvimento do sistema nervoso, em especial os que são importantes para o controle do apetite (e, portanto, do peso), o que não ocorria com os homens magros.
Barrès e seus colegas fizeram outra comparação sugestiva entre as marcações epigenéticas dos espermatozoides dos obesos antes da cirurgia de redução de estômago e as desses mesmos participantes após a operação. Resultado: depois da cirurgia, o padrão epigenético das células lembrava o de homens com peso normal.
“O mais importante a respeito dessas descobertas é sugerir que tais modificações podem ocorrer em células germinativas, ou seja, os óvulos e espermatozoides, e ser transmitidas para gerações seguintes”, diz o médico Licio Augusto Velloso, professor da Faculdade de Ciências Médicas da Universidade Estadual de Campinas (FCM-Unicamp), que estuda os mecanismos celulares e moleculares ligados à origem da obesidade e do diabetes. “Os estudos epigenéticos avançaram muito na última década e se espera que, num futuro não muito distante, o mapeamento de fatores ambientais e de seu impacto em diferentes aspectos da epigenética nos ajude a prevenir doenças importantes”, afirma Velloso.
Enxergar o excesso de peso pelo prisma epigenético pode trazer mais uma peça relevante para o quebra-cabeça da epidemia global de obesidade e de doenças metabólicas ligadas a ela. Historicamente associado à saúde e à fartura, o excesso de peso se tornou um problema de grandes proporções primeiro nos países ricos, mas hoje é cada vez mais comum em países mais pobres – a começar pelo Brasil, onde quase 60% da população adulta está acima do peso considerado saudável, conforme dados do Instituto Brasileiro de Geografia e Estatística (IBGE). Muitos países em desenvolvimento passaram rapidamente de um contexto em que a desnutrição era um problema grave para outro em que a obesidade é muito mais preocupante.
Geneticists tell us that somewhere between 1 and 5 percent of the genome of modern Europeans and Asians consists of DNA inherited from Neanderthals, our prehistoric cousins.
At Vanderbilt University, John Anthony Capra, an evolutionary genomics professor, has been combining high-powered computation and a medical records databank to learn what a Neanderthal heritage — even a fractional one — might mean for people today.
We spoke for two hours when Dr. Capra, 35, recently passed through New York City. An edited and condensed version of the conversation follows.
Q. Let’s begin with an indiscreet question. How did contemporary people come to have Neanderthal DNA on their genomes?
A. We hypothesize that roughly 50,000 years ago, when the ancestors of modern humans migrated out of Africa and into Eurasia, they encountered Neanderthals. Matings must have occurred then. And later.
One reason we deduce this is because the descendants of those who remained in Africa — present day Africans — don’t have Neanderthal DNA.
What does that mean for people who have it?
At my lab, we’ve been doing genetic testing on the blood samples of 28,000 patients at Vanderbilt and eight other medical centers across the country. Computers help us pinpoint where on the human genome this Neanderthal DNA is, and we run that against information from the patients’ anonymized medical records. We’re looking for associations.
What we’ve been finding is that Neanderthal DNA has a subtle influence on risk for disease. It affects our immune system and how we respond to different immune challenges. It affects our skin. You’re slightly more prone to a condition where you can get scaly lesions after extreme sun exposure. There’s an increased risk for blood clots and tobacco addiction.
To our surprise, it appears that some Neanderthal DNA can increase the risk for depression; however, there are other Neanderthal bits that decrease the risk. Roughly 1 to 2 percent of one’s risk for depression is determined by Neanderthal DNA. It all depends on where on the genome it’s located.
Was there ever an upside to having Neanderthal DNA?
It probably helped our ancestors survive in prehistoric Europe. When humans migrated into Eurasia, they encountered unfamiliar hazards and pathogens. By mating with Neanderthals, they gave their offspring needed defenses and immunities.
That trait for blood clotting helped wounds close up quickly. In the modern world, however, this trait means greater risk for stroke and pregnancy complications. What helped us then doesn’t necessarily now.
Did you say earlier that Neanderthal DNA increases susceptibility to nicotine addiction?
Yes. Neanderthal DNA can mean you’re more likely to get hooked on nicotine, even though there were no tobacco plants in archaic Europe.
We think this might be because there’s a bit of Neanderthal DNA right next to a human gene that’s a neurotransmitter implicated in a generalized risk for addiction. In this case and probably others, we think the Neanderthal bits on the genome may serve as switches that turn human genes on or off.
Aside from the Neanderthals, do we know if our ancestors mated with other hominids?
We think they did. Sometimes when we’re examining genomes, we can see the genetic afterimages of hominids who haven’t even been identified yet.
A few years ago, the Swedish geneticist Svante Paabo received an unusual fossilized bone fragment from Siberia. He extracted the DNA, sequenced it and realized it was neither human nor Neanderthal. What Paabo found was a previously unknown hominid he named Denisovan, after the cave where it had been discovered. It turned out that Denisovan DNA can be found on the genomes of modern Southeast Asians and New Guineans.
Have you long been interested in genetics?
Growing up, I was very interested in history, but I also loved computers. I ended up majoring in computer science at college and going to graduate school in it; however, during my first year in graduate school, I realized I wasn’t very motivated by the problems that computer scientists worked on.
Fortunately, around that time — the early 2000s — it was becoming clear that people with computational skills could have a big impact in biology and genetics. The human genome had just been mapped. What an accomplishment! We now had the code to what makes you, you, and me, me. I wanted to be part of that kind of work.
So I switched over to biology. And it was there that I heard about a new field where you used computation and genetics research to look back in time — evolutionary genomics.
There may be no written records from prehistory, but genomes are a living record. If we can find ways to read them, we can discover things we couldn’t know any other way.
Not long ago, the two top editors of The New England Journal of Medicine published an editorial questioning “data sharing,” a common practice where scientists recycle raw data other researchers have collected for their own studies. They labeled some of the recycling researchers, “data parasites.” How did you feel when you read that?
I was upset. The data sets we used were not originally collected to specifically study Neanderthal DNA in modern humans. Thousands of patients at Vanderbilt consented to have their blood and their medical records deposited in a “biobank” to find genetic diseases.
Three years ago, when I set up my lab at Vanderbilt, I saw the potential of the biobank for studying both genetic diseases and human evolution. I wrote special computer programs so that we could mine existing data for these purposes.
That’s not being a “parasite.” That’s moving knowledge forward. I suspect that most of the patients who contributed their information are pleased to see it used in a wider way.
What has been the response to your Neanderthal research since you published it last year in the journal Science?
Some of it’s very touching. People are interested in learning about where they came from. Some of it is a little silly. “I have a lot of hair on my legs — is that from Neanderthals?”
But I received racist inquiries, too. I got calls from all over the world from people who thought that since Africans didn’t interbreed with Neanderthals, this somehow justified their ideas of white superiority.
It was illogical. Actually, Neanderthal DNA is mostly bad for us — though that didn’t bother them.
As you do your studies, do you ever wonder about what the lives of the Neanderthals were like?
It’s hard not to. Genetics has taught us a tremendous amount about that, and there’s a lot of evidence that they were much more human than apelike.
They’ve gotten a bad rap. We tend to think of them as dumb and brutish. There’s no reason to believe that. Maybe those of us of European heritage should be thinking, “Let’s improve their standing in the popular imagination. They’re our ancestors, too.’”
Signatures of ethnicity in the genome appear to reflect an ethnic group’s shared culture and environment, rather than their common genetic ancestry, report scientists. Epigenetic signatures distinguishing Mexican and Puerto Rican children in this study cannot be explained by genetic ancestry alone, the researchers say.
A UC San Francisco-led study has identified signatures of ethnicity in the genome that appear to reflect an ethnic group’s shared culture and environment, rather than their common genetic ancestry.
The study examined DNA methylation — an “annotation” of DNA that alters gene expression without changing the genomic sequence itself — in a group of diverse Latino children. Methylation is one type of “epigenetic mark” that previous research has shown can be either inherited or altered by life experience. The researchers identified several hundred differences in methylation associated with either Mexican or Puerto Rican ethnicity, but discovered that only three-quarters of the epigenetic difference between the two ethnic subgroups could be accounted for by differences in the children’s genetic ancestry. The rest of the epigenetic differences, the authors suggest, may reflect a biological stamp made by the different experiences, practices, and environmental exposures distinct to the two ethnic subgroups.
The discovery could help scientists understand how social, cultural, and environmental factors interact with genetics to create differences in health outcomes between different ethnic populations, the authors say, and provides a counterpoint to long-standing efforts in the biomedical research community to replace imprecise racial and ethnic categorization with genetic tests to determine ancestry.
“These data suggest that the interplay between race and ethnicity as social constructs and genetic ancestry as a biological construct is more complex than we had realized,” said Noah Zaitlen, PhD, a UCSF assistant professor of medicine and co-senior author on the new study. “In a medical context both elements may provide valuable information.”
The research — published January 3, 2017 in the online journal eLife — was led by Joshua Galanter, MD, MAS, formerly an assistant professor of medicine, of bioengineering and therapeutic sciences, and of epidemiology and biostatistics at UCSF, who is now a scientist at Genentech. The research was jointly supervised by Zaitlen and co-senior author Esteban Burchard, MD, MPH, a professor of bioengineering and therapeutic sciences and of medicine in UCSF’s schools of Pharmacy and Medicine and the Harry Wm. and Diana V. Hind Distinguished Professorship in Pharmaceutical Sciences II at UCSF.
“This is a big advancement of our understanding of race and ethnicity,” Burchard said. “There’s this whole debate about whether race is fundamentally genetic or is just a social construct. To our knowledge this is the first time anyone has attempted to quantify the molecular signature of the non-genetic components of race and ethnicity. It demonstrates in a whole new way that race combines both genetics and environment.”
Teasing apart roles of genetics, environment in ethnic differences in disease
Researchers and clinicians have known for many years that different racial and ethnic populations get diseases at different rates, respond differently to medications, and show very different results on standard clinical tests: “For a whole range of medical tests, whether your physician is told that your lab result is normal or abnormal depends entirely on the race/ethnicity box that you tick on an intake form,” Zaitlen said.
It’s tempting to assume that such health disparities between races and ethnicities all stem from inherited genetic differences, but that’s not necessarily the case. Different racial and ethnic groups also eat different diets, live in neighborhoods with more or less pollution, experience different levels of poverty, and are more or less likely to smoke tobacco, all of which could also impact their health outcomes.
“A lot of our research involves trying to tease apart how much of health differences between populations are genetic and how much are environmental,” Zaitlen said.
The researchers turned to epigenetics to search for answers to these questions because these molecular annotations of the genetic code have a unique position between genetic ancestry and environmental influence. Unlike the rest of the genome, which is only inherited from an individual’s parents (with random mutations here and there), methylation and other epigenetic annotations can be modified based on experience. These modifications influence when and where particular genes are expressed and appear to have significant impacts on disease risk, suggesting explanations for how environmental factors such as maternal smoking during pregnancy can influence a child’s risk of later health problems.
Epigenetic signatures of ethnicity could be biomarkers for shared cultural experiences
In the new study, the team examined methylation signatures in 573 children of self-identified Mexican or Puerto-Rican identity drawn from the GALA II study, a cohort previously developed by Burchard to study environmental and genetic components of asthma risk in Latino children. They identified 916 methylation sites that varied with ethnic identity, but found that only 520 of these differences could be completely explained by genetic ancestry — 109 could be partially explained by ancestry, while 205 could not be explained by ancestry at all.
Overall, the researchers found that about 76 percent of the effect of ethnicity on DNA methylation could be accounted for by controlling for genetic ancestry, suggesting that nearly a quarter of the effect must be due to other, unknown factors. The researchers found that many of these additional methylation sites corresponded to sites that previous studies had shown to be sensitive to environmental and social factors such as maternal smoking, exposure to diesel exhaust, and psychosocial stress. This led the team to hypothesize that a large fraction of their newly disovered epigenetic markers of ethnicity likely reflect biological signatures of environmental, social, or cultural differences between ethnic subgroups.
“This suggests that using epigenetics as a biomarker could give you a lot of information about environmental exposures within particular populations that’s not captured by genetics,” Zaitlen said. “Our next step will be to understand how specific epigenetic signatures are linked to particular environmental exposures, and use those signals to understand patient risk.”
Scientists and clinicians have increasingly tried to move away from simplistic racial and ethnic categories in disease research, the authors say, and — with the rise of precision medicine — in clinical diagnosis and treatment as well. Studies by the Burchard group and others have found that using genetic ancestry rather than ethnic self-identification significantly improves diagnostic accuracy for certain diseases.
But the new data showing that a large fraction of epigenetic signatures of ethnicity reflect something other than ancestry suggests that abandoning the idea of race and ethnicity altogether could sacrifice a lot of valuable information about the drivers of differences in health and disease between different communities.
“Like a standard family history, ethnicity is association with disease for both genetic and environmental reasons,” Zaitlen said. “If your dad or mom had a heart attack, that tells doctors a lot about your risk for a heart attack. Part of that is genetic, but part of it is that your lifestyle is influenced heavily by your parents’ lifestyle. Your ethnic group is like a much bigger family — it’s partly a matter of genetics, but it also reflects the environment of your broader community.”
Journal Reference:
Joshua M Galanter, Christopher R Gignoux, Sam S Oh, Dara Torgerson, Maria Pino-Yanes, Neeta Thakur, Celeste Eng, Donglei Hu, Scott Huntsman, Harold J Farber, Pedro C Avila, Emerita Brigino-Buenaventura, Michael A LeNoir, Kelly Meade, Denise Serebrisky, William Rodríguez-Cintrón, Rajesh Kumar, Jose R Rodríguez-Santana, Max A Seibold, Luisa N Borrell, Esteban G Burchard, Noah Zaitlen. Differential methylation between ethnic sub-groups reflects the effect of genetic ancestry and environmental exposures. eLife, 2017; 6 DOI: 10.7554/eLife.20532
Imagem de reprodução de DNA de hélice quádrupla – Divulgação/Jean-Paul Rodriguez
WASHINGTON — Um grupo de cientistas propôs, nesta quinta-feira, um projeto ambicioso para criar um genoma humano sintético, que tornaria possível a criação de seres humanos sem a necessidade de pais biológicos. Esta possibilidade levanta polêmica sobre o quanto a vida humana pode ou deve ser manipulada.
O projeto, que surgiu em uma reunião de cientistas da Universidade Harvard, nos EUA, no mês passado, tem como objetivo desenvolver e testar o genoma sintético em células dentro de laboratório ao longo de dez anos. O genoma sintético humano envolve a utilização de produtos químicos para criar o DNA presente nos cromossomas humanos. A meta foi relatada na revista “Science” pelos 25 especialistas envolvidos.
Os cientistas propuseram lançar, ainda este ano, o que chamaram de Projeto de Escrita do Genoma Humano e afirmaram que iriam envolver o público nessa discussão, que incluiria questões éticas, legais e sociais.
Os especialistas esperam arrecadar US$ 100 milhões — o equivalente a R$ 361 milhões — em financiamento público e privado para lançar o projeto este ano. No entanto, eles consideram que os custos totais serão inferiores aos US$ 3 milhões utilizados no Projeto do Genoma Humano original, que mapeou pela primeira vez o DNA humano.
O novo projecto “incluirá a engenharia completa do genoma de linhas de células humanas e de outros organismos importantes para a agricultura e saúde pública, ou aqueles que interpretar as funções biológicas humanas”, escreveram na “Science” os 25 cientistas, liderados pelo geneticista Jef Boeke, do Centro Médico Langone, da Universidade de Nova York.







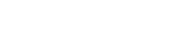

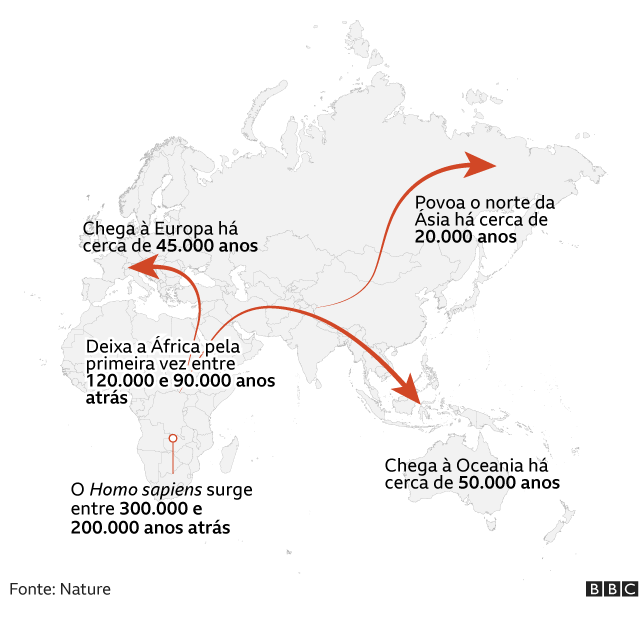

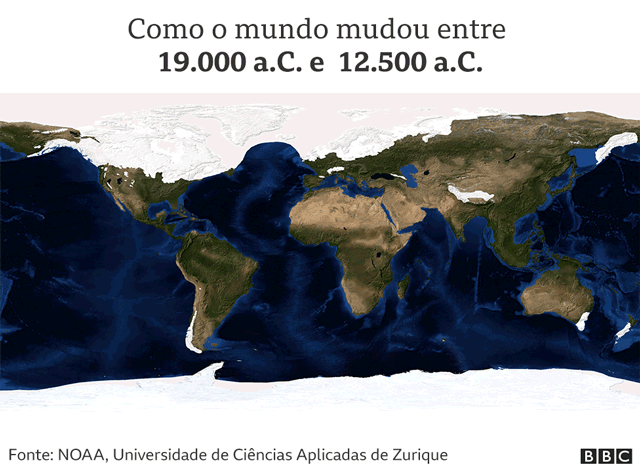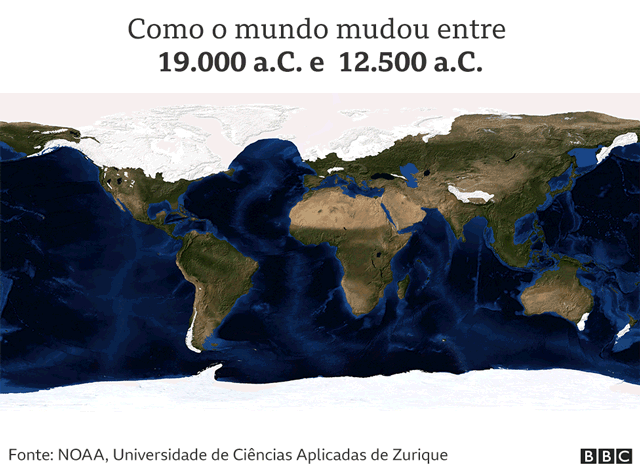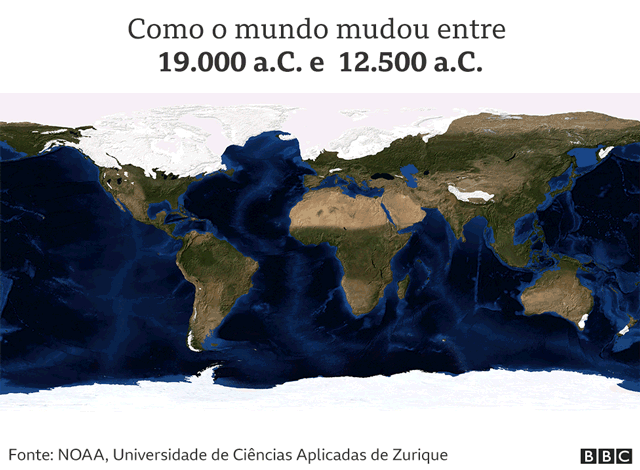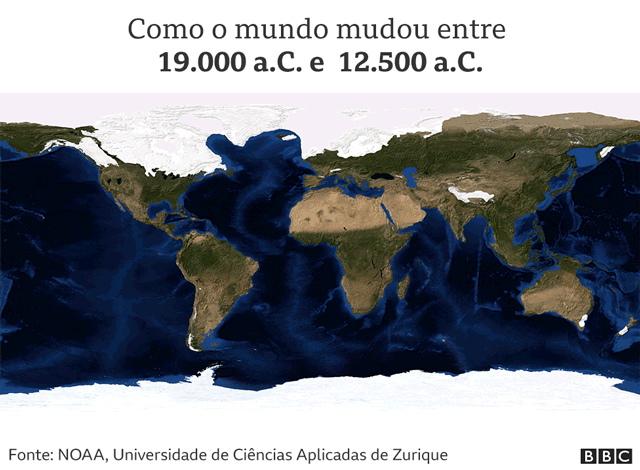
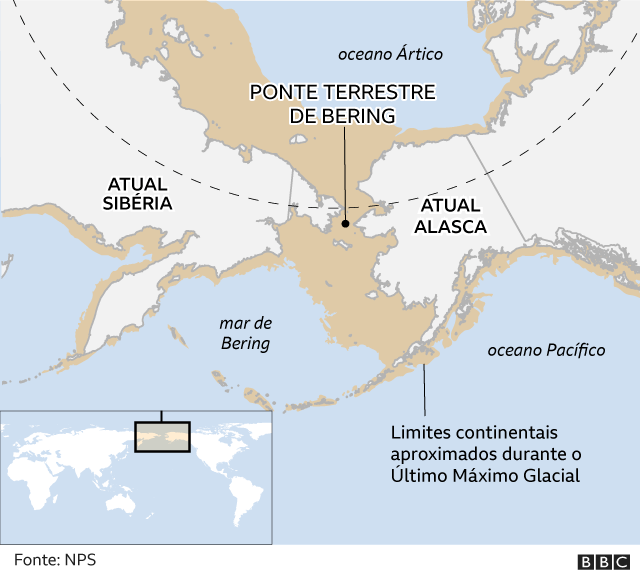


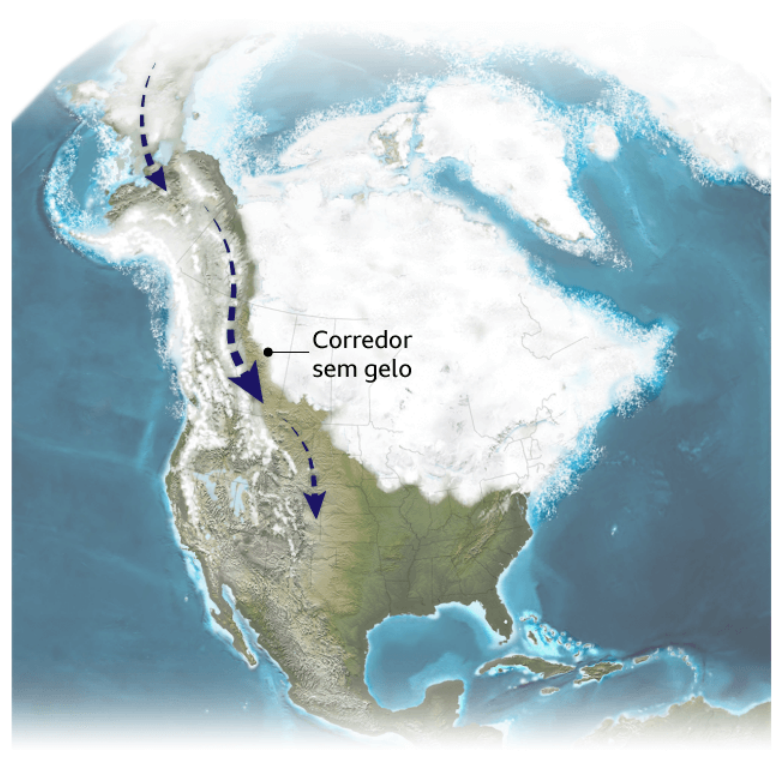

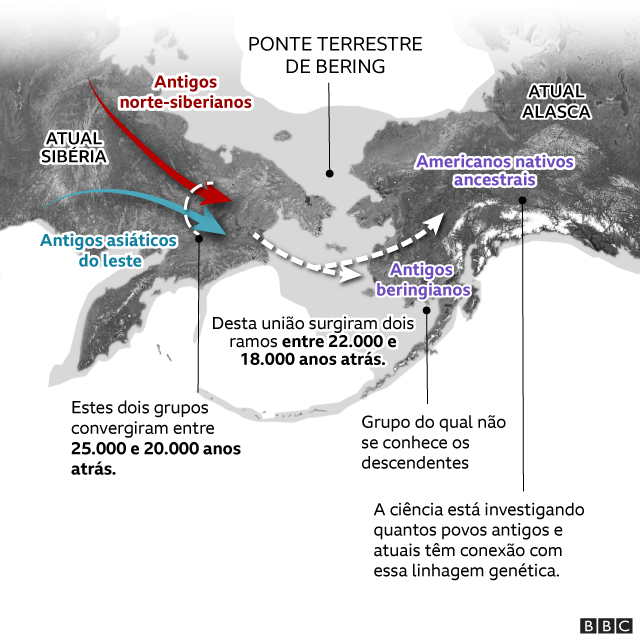































Você precisa fazer login para comentar.